



Las lesiones selares son heterogéneas en su naturaleza y abordaje. No todas requieren cirugía.
ObjetivosDescribir las formas de presentación de las lesiones selares y la presencia de endocrinopatías a lo largo de la evolución, así como resaltar la importancia de la valoración endocrinológica e identificar aquellas lesiones subsidiarias de tratamiento hormonal.
Pacientes y métodos Estudio retrospectivo de los niños menores de 14 años remitidos a nuestro centro por lesiones selares durante 12 años. Se recogieron las siguientes variables: sexo, edad, naturaleza de la lesión, presentación clínica, tamaño, tratamiento primario y presencia de endocrinopatías.
ResultadosSe incluyen 45 pacientes (25 mujeres) con edades comprendidas desde los 3 meses hasta 13,5 años (media 7,2±4,1) y un tiempo de seguimiento de 6,2±3,7 años. Se conoce la naturaleza de la lesión en 39 casos, de los cuales 4 se han tratado eficazmente por Endocrinología: 3 prolactinomas con cabergolina, y una hiperplasia hipofisaria con levotiroxina. El motivo de consulta fueron síntomas de neuropatía y oftalmopatía en 25/45 casos, y de endocrinopatía en 13/45. El periodo de síntomas endocrinológicos previos fue de 12,6±18,2 meses, frente a 2,6±4,9 meses de los neurooftalmológicos (p=0,012). En el momento del diagnóstico, 24/45 pacientes presentaban alguna endocrinopatía, ascendiendo a 37/45 al final del seguimiento.
ConclusionesLas lesiones del área selar requieren un abordaje multidisciplinario. El estudio endocrinológico es imprescindible para identificar las que son subsidiarias de tratamiento hormonal. Los síntomas o signos de endocrinopatía pueden aparecer antes que los de neuropatía u oftalmopatía, por lo que es fundamental reconocerlos para adelantar el diagnóstico. La evaluación hormonal debe repetirse periódicamente a lo largo de la evolución.
Sellar masses are an heterogeneous group of lesions, both in nature and management. Not all of them require surgery.
ObjectivesTo describe the presenting symptoms of sellar masses and endocrine abnormalities occurring during follow-up. To emphasize the significance of endocrine assessment, and to identify lesions amenable to hormonal treatment.
Patients and methodsA retrospective review of the records of children under 14 years of age referred to our center for sellar lesions during 12 years. Data collected included sex, age, nature of lesion, clinical presentation, size, treatment, and endocrine abnormalities.
ResultsForty-five patients (25 females) aged 7.2±4.1 years (range 0.25-13.5) were enrolled. Follow-up time was 6.2±3.7 years. Lesion nature was known in 39 cases, 4 of which were successfully treated at the Endocrinology Department: 3 prolactinomas (with dopamine agonist) and one thyrotroph cell hyperplasia (with levothyroxine). The most common presenting symptoms were neurological and/or visual (25/45), followed by endocrine conditions (13/45). Duration of endocrine and neuro-ophthalmic symptoms was 12.6±18.2 months and 2.6±4.9 (P=.012), respectively. Some endocrine condition was found in 24/45 patients at the initial evaluation and in 37/45 patients at the end of follow-up.
ConclusionsManagement of sellar lesions requires a multidisciplinary effort. Endocrine tests are indispensable to identify lesions amenable to hormonal treatment. Endocrine disorders usually occurred before neurological and ophthalmological symptoms, and their identification may therefore allow for earlier diagnosis. Hormone assessment should be regularly performed during follow-up.
Las lesiones del área selar (hipotálamo-hipofisaria-quiasmática) son infrecuentes en niños y adolescentes (solo un 6% de ellas aparecen en estas edades) y muy heterogéneas en su naturaleza y forma de presentación clínica1,2. La casuística es diferente a la del adulto3; su diagnóstico diferencial debe ser correcto, pues el abordaje terapéutico es muy distinto según la etiología.
El craneofaringioma es la lesión más frecuente en las 2 primeras décadas de la vida, y su tratamiento es quirúrgico. El prolactinoma es raro en estas edades, pero eso no quiere decir que no haya que contar con él, y su tratamiento es médico, así como el de la hiperplasia hipofisaria. Otras lesiones de esta área tampoco necesitan cirugía, tales como los tumores germinales, cuyo tratamiento de elección es la quimioterapia y la radioterapia, y los gliomas de las vías ópticas, que muchas veces no requieren ninguna actuación4–6.
Aunque la mayoría son histológicamente benignas, se asocian con una alta morbilidad por su proximidad a estructuras vitales. Suelen diagnosticarse por alteraciones visuales o neurológicas por efecto masa. También asocian endocrinopatías que incluyen enfermedad hipotalámica e hipofisaria por hipoproducción o por hiperproducción de alguna hormona. Con mucha frecuencia estas son las primeras manifestaciones de la lesión2,7.
Los objetivos de nuestro estudio son describir las formas de presentación de las lesiones del área selar en niños y adolescentes, analizar la presencia de endocrinopatías en el momento de su presentación y a lo largo de la evolución, y resaltar la importancia de la valoración endocrinológica, identificando aquellas subsidiarias de tratamiento hormonal.
Pacientes y métodosHemos realizado un estudio retrospectivo de los pacientes menores de 14 años remitidos a una Unidad de Endocrinología Pediátrica de un hospital terciario por la presencia de una masa selar durante un periodo de 12 años (2000-2011). Las lesiones habían sido diagnosticadas por resonancia magnética nuclear.
Para cada paciente se recogieron de las historias clínicas las siguientes variables: sexo, edad al diagnóstico, naturaleza de la lesión, síntomas de presentación clínica y su momento de inicio, diámetro del eje mayor de la masa al diagnóstico en la resonancia magnética nuclear, tratamiento primario y presencia de endocrinopatías al diagnóstico y a lo largo del periodo de seguimiento.
Los síntomas y signos de presentación clínica se definieron como los primeros relacionados con la lesión que llevaron a consultar, pero, además, se recogieron todos aquellos referidos u observados antes del diagnóstico. Se distinguieron distintas formas de presentación según estos fueran derivados de neuropatías (cefalea, signos de hipertensión craneal, convulsiones, signos focales), oftalmopatías (disminución de la agudeza o del campo visual, estrabismo, nistagmo) y endocrinopatías. El periodo de clínica previa se definió como el tiempo entre el inicio de los síntomas atribuibles a la lesión (aunque no llevaran a consultar) y el diagnóstico de la misma.
Como endocrinopatías se incluyeron las enfermedades de origen hipotalámico (obesidad, trastornos de la conducta alimentaria, de la sed, del sueño o de la temperatura) e hipofisario, ya sea por hipofunción de alguna de las hormonas de la hipófisis anterior (déficit de hormona del crecimiento [GH], hipocortisolismo, hipotiroidismo e hipogonadismo centrales) o de la posterior (diabetes insípida central), o por hiperproducción de las mismas (hiperprolactinemia, pubertad precoz y gigantismo). Para el diagnóstico de obesidad se utilizaron umbrales del índice de masa corporal específicos de cada edad y sexo. Las evaluaciones clínicas y analíticas fueron las habituales en el protocolo asistencial de estos pacientes y se realizaron inicialmente al diagnóstico de la lesión (excepto en 3casos, un craneofaringioma, un prolactinoma y un quiste dermoide, en los que se realizaron tras la exéresis quirúrgica), y posteriormente de forma semestral en todos los casos.
Las variables cuantitativas (edad y periodos de tiempo) se expresaron con sus medias±desviación típica. Los periodos de clínica previa neurooftalmológica y endocrinológica se compararon mediante la prueba estadística de Wilcoxon, y las proporciones de pacientes con endocrinopatía en cada grupo según la naturaleza de la lesión se compararon mediante la prueba de chi-cuadrado.
ResultadosSe incluyen en el estudio 45 pacientes (25 mujeres) con un rango de edad al diagnóstico de 3 meses a 13,5 años (media 7,2±4,1 años) y un tiempo de seguimiento de 6,2±3,7 años. En 29 casos el diámetro mayor de la lesión era superior a 20mm. Se conoce la naturaleza de la lesión en 39 casos: 13 gliomas de las vías ópticas (5 en pacientes diagnosticados previamente de neurofibromatosis tipo 1), 10 tumores germinales, 6 craneofaringiomas, 4 prolactinomas, 4 granulomas por histiocitosis, un quiste dermoide y una hiperplasia hipofisaria secundaria a hipotiroidismo primario congénito de diagnóstico muy tardío, a los 9 años de edad. En la tabla 1 se presentan las variables en el momento del diagnóstico en la cohorte completa y según la naturaleza de las lesiones.
Variables en el momento del diagnóstico de las lesiones selares en la cohorte completa y según su naturaleza
| Todos | Gliomas | Tumores germinales | Craneofaringiomas | Prolactinomas | Histiocitomas | Sin diagnóstico | |
| N | 45 | 13 | 10 | 6 | 4 | 4 | 6 |
| Mujeres | 25 | 6 | 10 | 1 | 2 | 1 | 3 |
| Edad al diagnóstico (años) | 7,2±4,1 | 2,8±2,3 | 9,4±1,7 | 7,2±3,3 | 13,2±0,3 | 7,0±4,1 | |
| Meses previos con síntomas de endocrinopatía | 12,6±18,2 | 7,0±4,6 | 25,0±26,2 | 9,6±10,0 | 0,7±0,3 | 4,2±2,1 | |
| Meses previos con síntomas de neuropatía u oftalmopatía | 2,7±4,9 | 1,1±1,3 | 2,0±2,1 | 1,5±1,0 | 15,0±12,7 | ||
| Años de seguimiento | 6,2±3,7 | 7,9±3,3 | 5,5±3,0 | 7,1±5,1 | 3,2±2,0 | 5,5±5,9 | 6,0±2,4 |
| Forma de presentación | |||||||
| Clínica de neurooftalmopatía | 25 | 9 | 7 | 6 | 2 | 0 | 0 |
| Subclínica (neuroimagen) | 7 | 2 | 0 | 0 | 0 | 0 | 5 |
| Clínica de endocrinopatía | 24 | 3 | 10 | 3 | 2 | 4 | 1 |
| Como motivo de consulta | 13 | 2 | 3 | 0 | 2 | 4 | 1 |
| En la anamnesis dirigida | 11 | 1 | 7 | 3 | 0 | 0 | 0 |
| Pubertad precoz | 4 | 3 | 0 | 0 | 0 | 0 | 1 |
| Diabetes insípida | 14 | 0 | 9 | 2 | 0 | 3 | 0 |
| Galactorrea | 2 | 0 | 0 | 0 | 2 | 0 | 0 |
| Obesidad | 2 | 0 | 0 | 1 | 0 | 1 | 0 |
| Talla baja | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| Anorexia | 1 | 0 | 1 | 0 | 0 | 0 | 0 |
| Diámetro>20mm neuroimagen | 1 | 0 | 7 | 6 | 2 | 0 | 0 |
| Tratamiento de la lesión selar | |||||||
| Cirugía | 9 | 2 | 0 | 5 | 1 | 0 | 0 |
| Quimioterapia | 6 | 3 | 0 | 0 | 0 | 3 | 0 |
| Quimioterapia y radioterapia | 1 | 1 | 10 | 0 | 0 | 0 | 0 |
| Cirugía y quimioterapia | 1 | 1 | 0 | 0 | 0 | 0 | 0 |
| Cirugía y radioterapia | 1 | 0 | 0 | 1 | 0 | 0 | 0 |
| Cabergolina | 3 | 0 | 0 | 0 | 3 | 0 | 0 |
| Levotiroxina | 1 | ||||||
| Ninguno | 13 | 6 | 0 | 0 | 0 | 1 | 6 |
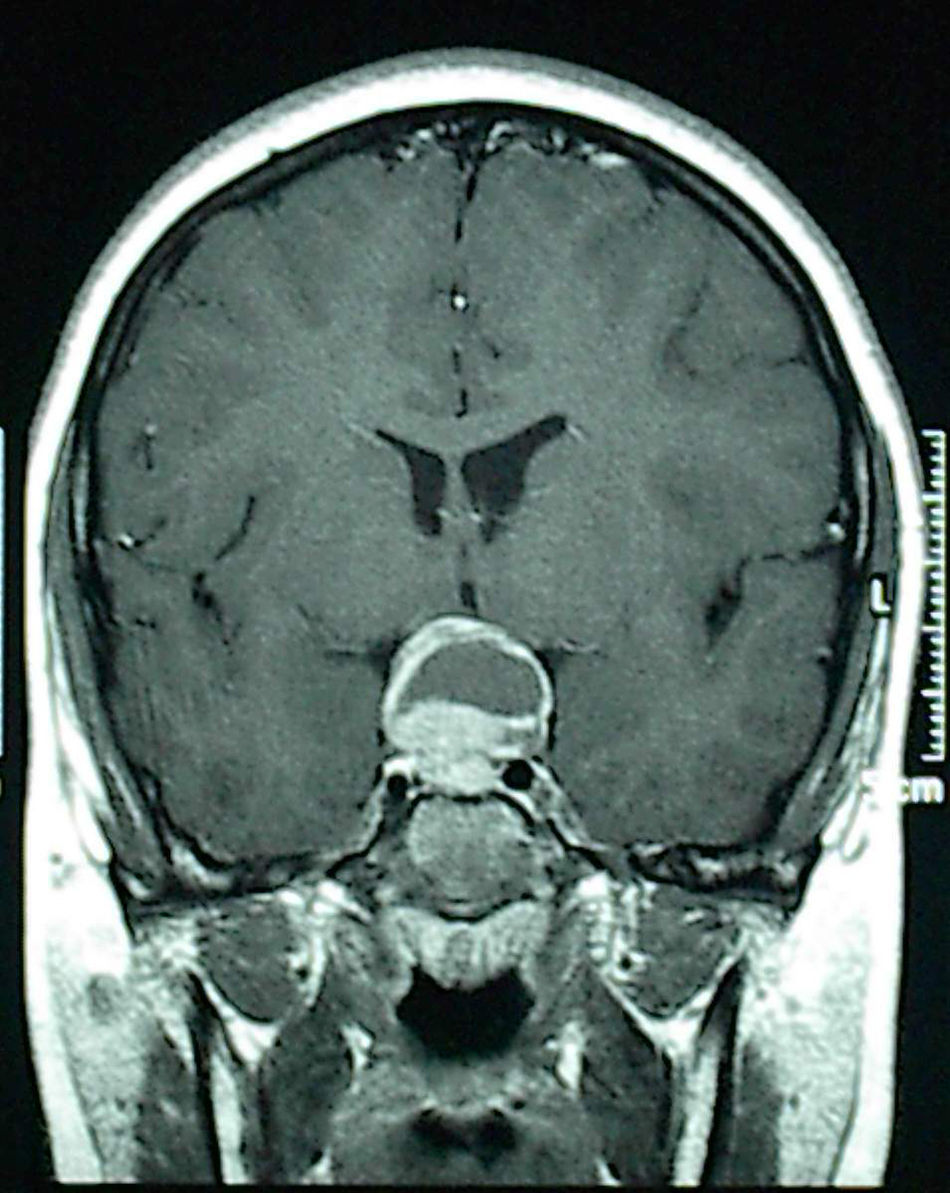
Cuatro casos se han tratado eficazmente por Endocrinología: 3 prolactinomas con el agonista dopaminérgico cabergolina, y la hiperplasia hipofisaria con levotiroxina. Un varón de 13 años que consultó por cefalea fue intervenido sin realizar estudio hormonal previo al presentar en la neuroimagen una lesión sospechosa de craneofaringioma por su aspecto quístico y gran extensión supraselar (fig. 1). Por Anatomía Patológica se diagnosticó de prolactinoma y el área quística era debida a apoplejía intratumoral. Los 3 pacientes con prolactinomas tratados médicamente, todos de 13 años de edad, fueron 2 mujeres púberes que consultaron por galactorrea aislada y presentaban un adenoma menor de 10mm, y un varón que consultaba por cefalea de meses de evolución y presentaba otro macroadenoma quístico. En la anamnesis de los varones no había ningún dato de endocrinopatía que pudiera haber adelantado el diagnóstico: no habían iniciado la pubertad y uno de ellos presenta una ligera ginecomastia de 15mm de diámetro bilateral, circunstancias que podían considerarse fisiológicas en esta edad.

Además de los 4 casos de tratamiento exclusivamente hormonal, en 17 niños el manejo fue oncológico (quimioterapia y/o radioterapia) y en 13 no se requirió ninguna terapia para la lesión (no presentaban manifestaciones clínicas y se mantenían estables según los controles periódicos de neuroimagen). Solamente 11 pacientes necesitaron cirugía de la masa, 2 de ellos seguida de quimioterapia y radioterapia. El estudio hormonal también descubrió 4 tumores secretores de gonadotropina coriónica; se estableció así el diagnóstico de germinoma sin necesidad de biopsia y se remitió a estos pacientes directamente a Oncología sin pasar por quirófano.
El motivo de consulta fueron síntomas relacionados con neuropatías y oftalmopatías en 25 casos y con endocrinopatías solo en 13: diabetes insípida en 6, pubertad precoz en 3, galactorrea en 2, hipocrecimiento en uno y ganancia ponderal en uno. Se diagnosticaron de forma subclínica por neuroimagen 7 lesiones: 2 en el estudio de neurofibromatosis tipo 1, y 5 que pueden considerarse incidentalomas, pues se investigaban problemas (epilepsia, retraso psicomotor, síncope) no relacionados con enfermedad selar. Hay que destacar que en 11 casos que consultaron por clínica neurológica o visual, en la anamnesis dirigida referían clínica endocrinológica de larga evolución no valorada hasta ese momento (8 de ellos polidipsia/poliuria abundante desde hacía uno a 5 años). El periodo medio de síntomas endocrinológicos previos fue de 12,6±18,2 meses, más largo que el de síntomas neurooftalmológicos, de 2,6±4,9 meses (p=0,012).
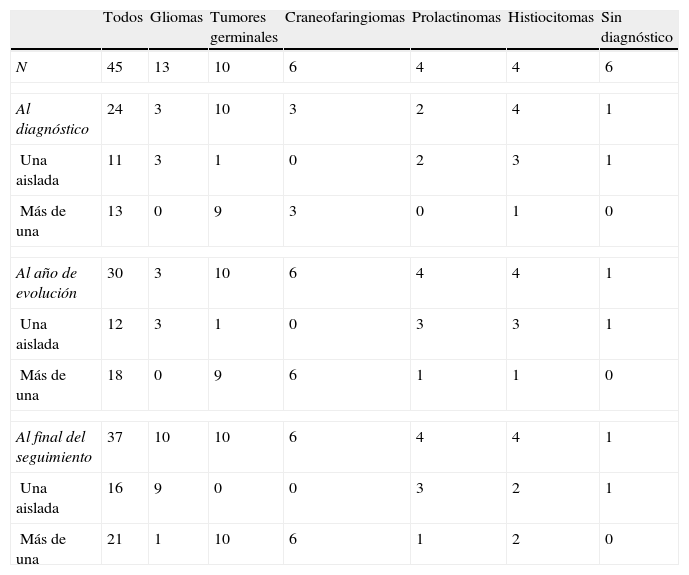
De los 24 pacientes que presentaban trastornos endocrinológicos en el momento del diagnóstico (los 13 en los que fueron motivo de consulta y los 11 en los que se descubrieron por la anamnesis dirigida), la mitad de ellos presentaban más de uno. La proporción de pacientes con alguna alteración hormonal ascendía a 30/45 al año de evolución, y a 37/45 al final del periodo de seguimiento (tabla 2). La presencia de endocrinopatías al diagnóstico y en el primer año de evolución se relacionó con la naturaleza de la lesión, pero con ninguna otra variable (edad, sexo, tamaño tumoral): los gliomas de las vías ópticas tienen menor proporción de alteraciones endocrinas al diagnóstico (p=0,004) y al año de evolución (p<0,001), 3/13 en ambas evaluaciones, que consistían todos ellos en pubertad precoz. La diferencia deja de ser significativa al final del periodo de seguimiento, donde 10/13 gliomas las presentan (p=0,62). Las 7 que se desarrollan a lo largo de la evolución lo hacen en un rango de 3 a 9 años (5 casos de pubertad precoz, uno de hipotiroidismo central y otro de diabetes insípida central). En todos los pacientes se trataba de endocrinopatías aisladas, salvo uno, el único que había recibido radioterapia, que asoció 2: pubertad precoz e hipotiroidismo centrales, a los 8 y 9 años de evolución, respectivamente.
Presencia de endocrinopatías en el momento del diagnóstico de las lesiones selares, al año de evolución y al final de periodo de seguimiento en la cohorte completa y según la naturaleza de las lesiones
| Todos | Gliomas | Tumores germinales | Craneofaringiomas | Prolactinomas | Histiocitomas | Sin diagnóstico | |
| N | 45 | 13 | 10 | 6 | 4 | 4 | 6 |
| Al diagnóstico | 24 | 3 | 10 | 3 | 2 | 4 | 1 |
| Una aislada | 11 | 3 | 1 | 0 | 2 | 3 | 1 |
| Más de una | 13 | 0 | 9 | 3 | 0 | 1 | 0 |
| Al año de evolución | 30 | 3 | 10 | 6 | 4 | 4 | 1 |
| Una aislada | 12 | 3 | 1 | 0 | 3 | 3 | 1 |
| Más de una | 18 | 0 | 9 | 6 | 1 | 1 | 0 |
| Al final del seguimiento | 37 | 10 | 10 | 6 | 4 | 4 | 1 |
| Una aislada | 16 | 9 | 0 | 0 | 3 | 2 | 1 |
| Más de una | 21 | 1 | 10 | 6 | 1 | 2 | 0 |
Nueve de 10 tumores germinales presentaban déficit completo anterohipofisario y posterohipofisario; todas estas lesiones eran mayores de 20mm en la neuroimagen en el momento del diagnóstico. Solo un caso, que consultó por diabetes insípida aislada y cuya neuroimagen mostraba un ensanchamiento del tallo hipofisario, desarrolló el déficit anterior 3 años más tarde, coincidiendo con el crecimiento del tumor, antes de iniciar el tratamiento quimioterápico y radioterápico. Tres de los 6 craneofaringiomas presentaban al diagnóstico panhipopituitarismo (incluyendo diabetes insípida central). Los otros 3 lo desarrollaron inmediatamente tras la cirugía (fig. 2)
Discusión
Nuestros datos enfatizan la importancia de identificar adecuadamente las lesiones selares subsidiarias de tratamiento hormonal en niños y adolescentes, así como de reconocer los síntomas y signos de endocrinopatía para adelantar el diagnóstico y manejarlas adecuadamente durante toda su evolución.
Aunque los adenomas sean raros en niños, es obligado descartar la presencia de un prolactinoma, pues responde a tratamiento médico. Sobre todo lo vamos a encontrar en mayores de 12 años, pero se ha descrito también en prepúberes. Una simple analítica basal con una prolactina mayor de 200ng/ml lo diagnostica fácilmente, por lo que ante cualquier masa selar o paraselar, incluso en prepúberes, e independientemente de su aspecto radiológico, debe determinarse antes de plantear el tratamiento quirúrgico2–5,8,9. Los adenomas pueden confundirse con craneofaringiomas, pues estos pueden tener también un origen intraselar. Aunque los primeros suelen ser sólidos y los segundos quísticos, los adenomas de larga evolución pueden crecer y presentar zonas quísticas por apoplejía (necrosis o hemorragia) intratumoral10–12. En nuestra serie, mientras que las mujeres púberes se diagnosticaron rápidamente por presentar galactorrea, en los varones no hubo síntomas de hipogonadismo (pues el retraso puberal solo se considera patológico a partir de la edad de 14 años), consultaron por cefalea, y presentaban al inicio grandes macroadenomas con zonas quísticas.
Los agonistas dopaminérgicos, sobre todo la cabergolina, son la primera elección en el tratamiento del prolactinoma; consiguen una disminución significativa del tamaño tumoral en más de la mitad de los casos, con normalización del nivel de prolactina y restauración de la función gonadal en más de tres cuartas partes, preservando el resto de la función hipofisaria. La cirugía queda, pues, reservada a situaciones de intolerancia o resistencia a la medicación8,9,13,14. La hiperplasia hipofisaria o seudoadenoma reactivo al hipotiroidismo primario de larga evolución es otro ejemplo de masa selar que se resuelve con tratamiento hormonal, tal y como hemos visto en uno de nuestros niños15.
Otra hormona imprescindible de analizar antes de plantear el manejo de la lesión es la gonadotropina coriónica. Casi la mitad de los tumores germinales hipotalámicos la producen, junto con la alfafetoproteína. En estas neoplasias el tratamiento de elección es quimioterapia y radioterapia, así que la presencia de estos marcadores hace innecesaria la biopsia. Como hemos visto en nuestra serie, suelen producir hipopituitarismo precozmente, sobre todo diabetes insípida central, frecuentemente acompañada de déficits de la hipófisis anterior. Los pocos casos que no presentan panhipopituitarismo al diagnóstico lo desarrollan tras el tratamiento oncológico16–18.
El craneofaringioma suele diagnosticarse por síntomas neurológicos, si bien el 80% de los pacientes presentan déficits hipofisarios que no suelen ser el motivo de consulta. Tras el tratamiento quirúrgico, prácticamente todos terminan de completar el panhipopituitarismo1,2,5,19–22. Como vemos, la mayoría de las lesiones selares presentan clínica endocrinológica al diagnóstico, y muchas de ellas, de larga evolución. En una amplia serie de 176 pacientes recientemente publicada, dos tercios referían esta clínica en un intervalo medio de 6 meses previo al inicio con síntomas de compromiso de espacio7. Las excepciones las suponen los gliomas y los granulomas de la histiocitosis, donde la afectación endocrina suele ser más secuencial, y los incidentalomas que son clínicamente silentes. En nuestros pacientes es llamativa la presencia de numerosos casos con diabetes insípida central de muy larga evolución (entre uno y 5 años) que no habían sido derivados para estudio por sus médicos, limitándose a descartar diabetes mellitus y trastornos renales o electrolíticos.
Los gliomas de las vías ópticas son los tumores que se diagnostican en edades más tempranas, sobre todo aquellos (casi la mitad de ellos) que aparecen en el contexto de una neurofibromatosis tipo 1. Suelen ser astrocitomas pilocíticos de bajo grado de malignidad que muchas veces no progresan e incluso involucionan. No presentan, generalmente, endocrinopatías al inicio, salvo los casos no diagnosticados previamente que consultan por pubertad precoz central o ganancia ponderal, pero sin déficits hipofisarios. Sin embargo, podrán desarrollarlos evolutivamente, con más probabilidad si han sido tratados con cirugía o radioterapia, pero incluso sin haber recibido ningún tratamiento23–25. Se ha descrito también en estos tumores hipercrecimiento por hipersecreción de GH26. En el seguimiento de nuestros niños, el hipopituitarismo se relaciona con haber recibido tratamiento primario del tumor, mientras que la pubertad precoz aparece también en no tratados.
La cuarta parte de los pacientes menores de 18 años con histiocitosis tienen déficits hipofisarios en sus 10 primeros años de evolución. Casi todos tienen diabetes insípida y la mitad de ellos asocian déficit de GH y, en menor proporción, de otras hormonas. La edad media de inicio de la histiocitosis es de 2,8 años, de la diabetes insípida de 3,9 años, y del déficit de GH de 7,7 años. Las afectaciones multiorgánica y craneofacial se asocian a la hipotalámica, y el tratamiento sistémico no las previene27–29.
Cada vez con más frecuencia encontramos en nuestros pacientes incidentalomas. Son masas insospechadas que se descubren casualmente en el estudio de otras enfermedades no relacionadas. Para su manejo contamos únicamente con guías de práctica clínica de adultos. Solo requieren cirugía aquellos que produzcan signos neurológicos o visuales, y los hipersecretores (salvo de prolactina); los demás, se vigilan con resonancia magnética nuclear semestral30.
Como vemos, las lesiones del área selar exigen un abordaje multidisciplinario. El estudio endocrinológico es imprescindible, puesto que el endocrinólogo puede tratar y resolver algunas de estas lesiones y sus consecuencias (prolactinomas, hiperplasias reactivas al hipotiroidismo, otros déficits hormonales), o simplificar el diagnóstico de otras (tumores germinales secretantes de gonadotropina coriónica). Es fundamental reconocer los síntomas de endocrinopatía para adelantar su diagnóstico, pues muchos de estos pacientes los presentan en el momento del inicio, bastante antes que los síntomas neurológicos y visuales. El seguimiento endocrinológico debe mantenerse en todos los casos, puesto que otras alteraciones hormonales van a ir presentándose a lo largo de la evolución.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
