


La diabetes tipo 1 se asocia a otras enfermedades autoinmunes, con más frecuencia a tiroiditis linfocitaria o a celiaquía. En la última década se ha descrito algunos casos de encefalitis límbica asociados a diabetes tipo 1, tiroiditis u otros procesos autoinmunes, incluso en edad pediátrica1–5. La encefalitis límbica es un proceso inflamatorio autoinmune que afecta a hipocampo y amígdala, que hasta hace pocos años se consideraba de origen exclusivamente paraneoplásico. Los pacientes tienen en sangre y/o LCR anticuerpos (Ac.) contra la superficie neuronal o contra antígenos intracelulares, como los Ac. anti-GAD. El GAD, que se expresa selectivamente en las neuronas y en las células β pancreáticas, es el enzima que limita la tasa de síntesis de GABA (ácido γ aminobutírico), principal neurotransmisor inhibitorio que modula y sincroniza la actividad neuronal en el SNC. Estos Ac. inhiben la actividad del GAD, posiblemente mediados por linfocitos T citotóxicos, y reducen la síntesis o exocitosis de GABA. Originan alteraciones neuropsiquiátricas progresivas como alucinaciones, epilepsia temporal refractaria, disfunción de la memoria y deterioro cognitivo, que pueden revertir con tratamiento inmunomodulador4–6.
Un paciente tiene el «síndrome límbico» si cumple más de uno de los siguientes criterios: alteración reciente de la memoria, crisis de lóbulo temporal, anomalías psiquiátricas, y además tener más de uno de estos: neuropatología (encefalitis temporo medial crónica); tumor diagnosticado en los siguientes 5 años del inicio de los síntomas y signos neurológicos; anticuerpos onconeuronales o Ac. anti-VGKC, NMDAR, GAD; hallazgo en la resonancia cerebral de un inexplicado incremento de señal temporo medial FLAIR/T26–9.
Se presenta el caso de una niña que comenzó con diabetes mellitus tipo 1 con 4 años, y que a los 7 comenzó con deterioro cognitivo y trastorno de conducta. En su historia no había antecedentes familiares ni personales de interés. El desarrollo psicomotor había sido normal hasta los 7 años, pero a partir de entonces refieren trastornos progresivos de conducta (agresividad, desobediencia), pérdida de memoria, mal rendimiento académico, miedo irracional, angustia, alucinaciones auditivas, trastorno del sueño y crisis de ausencias. Estas aumentaron en intensidad y frecuencia mostrando semiología de crisis parciales complejas. A los 10 años tenía muchos episodios diarios consistentes en clonías faciales, versión ocular, hipertonía y sacudidas tónico clónicas generalizadas. Simultáneamente empeoraba el deterioro cognitivo y el rendimiento escolar, destacando la disfunción ejecutiva y la falta de memoria. También tenía escaso control emocional y de impulsos, trastornos graves de conducta y agresividad (pegaba a los compañeros, se desnudaba en clase), precisando un colegio de educación especial.
A pesar del tratamiento intensivo de la diabetes con insulina detemir y lispro en pauta basal-bolus y raciones, se controlaba mal por las numerosas crisis diarias que producían hiperglucemias e hipoglucemias; HbA1c 7,8%, TSH 3.00, T4L 0,79, microalbuminuria negativa, Ac. antitiroideos negativos. Ac. anti-transglutaminasa IgA negativos. Había presentado, también, pubertad adelantada que se trató con triptorelina.
Dada la refractariedad de sus crisis epilépticas, a los 12 años fue remitida a un hospital terciario para valorar el tratamiento quirúrgico de la epilepsia, donde se realizó:
- –
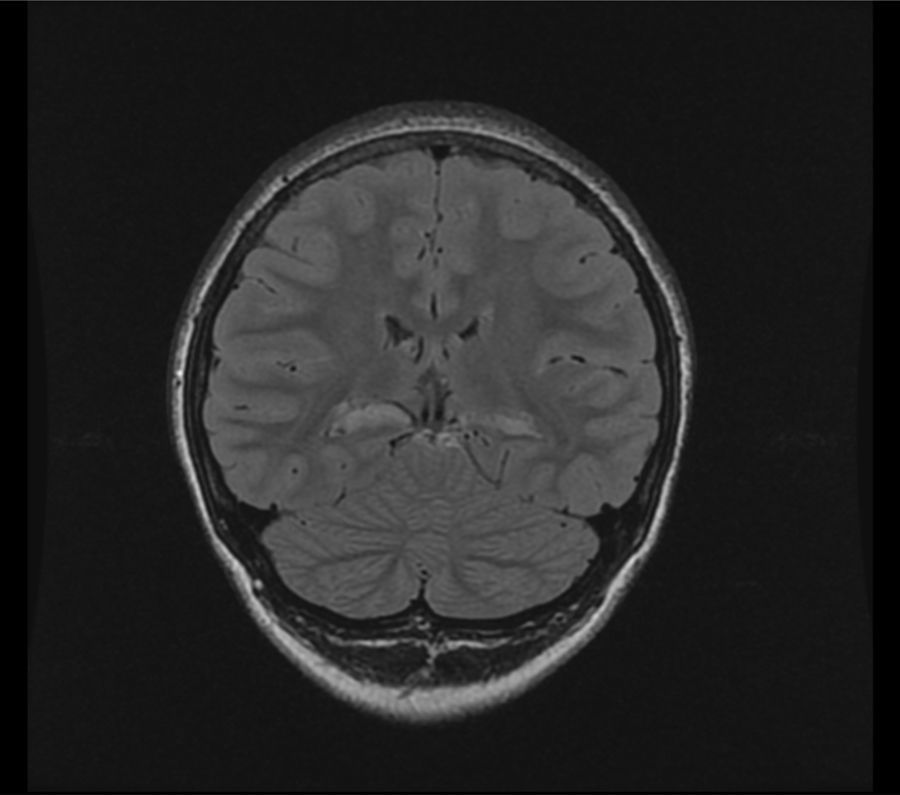
RM 3T: atrofia de ambos hipocampos, con aumento de señal en T2 y FLAIR, que sugieren esclerosis temporal medial bilateral. Mayor disminución del volumen cabeza hipocampo izquierdo, los cambios de señal son más severos en el derecho (fig. 1).
- –
PET/TAC: hipometabolismo temporal bilateral, más marcado y extenso en lóbulo temporal derecho, que afecta región mesial, polo temporal y neocórtex.
- –
Video EEG: anomalías intercríticas: anomalías epileptiformes temporal derecha e izquierda: 26 crisis temporales derechas y 4 tónico-clónicas generalizadas.
- –
Valoración neuropsicológica: deterioro cognitivo/evolutivo en todos los dominios, cociente intelectual previo 69, actual 51), alteraciones de memoria, sobre todo verbal. Disfunción ejecutiva, dificultades para control emocional y de impulsos, grave trastorno del comportamiento, conducta disocial.

Se catalogó como epilepsia refractaria con esclerosis temporal medial bilateral con predominio derecho, refractaria al tratamiento farmacológico y se realizó una lobectomía temporal derecha con amígdalo-hipocampectomía derecha.
A los 3 meses de la neurocirugía mejoraron las crisis, pero empeoró el cuadro psicótico esquizofrenia-like, y se reevaluó la etiología:
- –
En la biopsia operatoria se observaba neuronofagía y con tinción CD3 para linfocitos aparecían «nódulos microgliales» de linfocitos, ambos compatibles con encefalitis. Se estudiaron los anticuerpos de encefalopatía autoinmune que resultaron negativos (NMDA, AMPA, GABA, mGLuR1, mGLUR5, VGKC y anti-GAD). Bandas oligoclonales en LCR: positivas. Ecografía abdominal: normal.
- –
Ante los hallazgos histológicos compatibles con encefalitis, la enfermedad endocrinológica autoinmune (diabetes tipo 1) y las bandas oligoclonales en LCR, se sospechó de «encefalitis autoinmune» y se pautó inmunoterapia. Se inició tratamiento inmunomodulador con megadosis de metilprednisolona y ciclos mensuales de inmunoglobulina IV (2g/kg/ciclo), observando ya desde el tercero, mejoría franca de la escolaridad (memoria, lenguaje y cálculo) y la conducta, desapareciendo las crisis.
- –
A los 3 años de la inmunoterapia, con 15 de edad no ha necesitado más tratamiento, ni ha tenido otras enfermedades. Muestra buena integración familiar y escolar, sin alteraciones del comportamiento. La evolución cognitiva es favorable con mejoría del rendimiento escolar, lenguaje, atención y memoria. Desaparecieron las alucinaciones auditivas. No tiene crisis, pero toma oxcarbamazepina por anomalías esporádicas del EEG. El control de su diabetes mejoró con la misma pauta: alimentación por raciones e insulina basal-bolus con detemir y lispro.
- –
Exploración física normal, peso 51kg (−0,61DE); talla 151cm (−1,78DE); Tanner V; HbA1C 6,7%; TSH 3,1mcUI/ml; T4L 0,90ng/dl; colesterol 176mg/dl; ACTH 43,8pg/ml; cortisol 22μg/dl y Ac. anti-transglutaminidasa negativos. Microalbuminuria negativa y Ac. antitiroideos negativos.
Este caso es interesante por la reciente identificación del origen autoinmune en el síndrome límbico y su rareza entre la población pediátrica3,4. Se ha descrito que los diabéticos con Ac. anti-GAD tienen mayor riesgo de deterioro neurológico, por el papel del GAD en la síntesis del GABA (especialmente con niveles altos)5,10. La síntesis intratecal de Ac. puede persistir mucho tiempo, originando el cuadro clínico años después de su detección en sangre6, y no siempre se detectan4. Se presentan en edades más jóvenes que otros tipos de encefalitis autoinmune, y parecen tener mejor respuesta al tratamiento que la originada por otros Ac. anti-antígenos intracelulares6. También es interesante por la dificultad del diagnóstico, al que se llegó tras lobectomía temporal derecha, indicada por epilepsia refractaria con deterioro neurocognitivo. Precisamente por esta rareza y novedad, solo se sospechó el origen autoinmune tras los hallazgos histológicos compatibles con encefalitis, confirmándose al completar el estudio. Destaca especialmente la evolución clínica de esta niña, que tras años de crisis y progresivo deterioro neurológico, cognitivo y conductual, y sin mejorar con la neurocirugía, tuvo una excelente respuesta a las inmunoglobulinas, sin recaídas, ni efectos secundarios. Pensamos que este caso puede contribuir a identificar y diagnosticar en el futuro otros similares, evitar la neurocirugía y facilitar que se utilicen más precozmente las inmunoglobulinas dada su eficacia4,6–8.
