



La apoplejía hipofisaria (AH) clásica es un síndrome clínico agudo, potencialmente fatal, provocado por la hemorragia y/o infarto de la glándula hipofisaria. Constituye uno de los cuadros clínicos de urgencia neuroendocrinológica. Sin embargo, no existe un consenso claro sobre cuáles pueden ser las mejores opciones para su diagnóstico y tratamiento.
ObjetivoElaborar una guía de práctica clínica con una serie de recomendaciones para el diagnóstico y tratamiento de los pacientes con AH basadas en la evidencia médica disponible, que sirva de ayuda a los profesionales implicados en su cuidado.
MétodosSe ha tomado como base la guía clínica de diagnóstico y tratamiento de la apoplejía hipofisaria, publicada en el año 2006 por el Grupo de Trabajo de Neuroendocrinología de la Sociedad Española de Endocrinología y Nutrición (SEEN), así como la Guía Británica de Práctica Clínica publicada en 2011. Se ha trabajado en la adaptación al formato utilizado en la mayoría de las revistas médicas internacionales. Para ello, tras la revisión bibliográfica actualizada, se ha evaluado la calidad de la evidencia y el peso de las recomendaciones de acuerdo con el sistema propuesto por la Agency for Health Care Policy and Research (AHCPR).
ConclusionesEl diagnóstico de apoplejía hipofisaria debería valorarse en aquellos pacientes con cefalea aguda grave, con o sin síntomas neurooftalmológicos. Se realizará una evaluación clínica completa. La evaluación analítica urgente incluirá medición de electrólitos, función renal y hepática, hemograma y estudio de coagulación y concentraciones basales de hormonas hipofisarias y periféricas. Será crucial descartar el déficit de hormona adrenocorticotropa (ACTH). Si el estado del paciente lo permite, debe realizarse una campimetría y una resonancia magnética (RM) urgente para confirmar el diagnóstico. En pacientes con inestabilidad hemodinámica, disminución de nivel de conciencia, disminución de la agudeza visual y defectos extensos en el campo visual se recomienda realizar descompresión quirúrgica en la primera semana tras el inicio de los síntomas. Los pacientes con sintomatología más leve pueden ser tratados de forma conservadora, bajo supervisión estrecha. El tratamiento de la AH debería realizarse en un centro donde exista disponibilidad y experiencia en el tratamiento neuroquirúrgico por vía transesfenoidal y posibilidad de seguimiento oftalmológico y endocrinológico.
Classic pituitary apoplexy (PA) is an acute, life-threatening clinical syndrome caused by acute hemorrhage and/or infarction of the pituitary gland. PA is considered a neuroendocrinological emergency. However, there is no consensus about the best options for PA diagnosis and management.
ObjectiveTo develop a clinical practice guideline with a number of recommendations for diagnosis and treatment of patients with PA based on the medical evidence available, in order to help clinicians involved in their care.
MethodsThe clinical guideline for diagnosis and treatment of pituitary apoplexy issued in 2006 by the Neuroendocrinology Working Group of the Spanish Society of Endocrinology and Nutrition (SEEN) and the British Clinical Practice Guideline published in 2011 were taken as the basis. The text has been adapted to the format used in most international medical journals. For this, after updated medical literature, the quality of evidence and the strength of the recommendations were evaluated using the system proposed by the Agency for Health Care Policy and Research (AHCPR).
ConclusionsDiagnosis of pituitary apoplexy should be considered in all patients with acute severe headache with or without neuro-ophthalmic signs. Patients with PA must undergo a complete history and physical examination. All patients with suspected pituitary apoplexy should have urgent blood samples drawn to test electrolytes, renal function, liver function, coagulation screen, complete blood count, and basal levels of pituitary and peripheral hormones, and to rule out adrenocorticotropic hormone (ACTH) deficiency. Formal visual field assessment should be performed when the patient is clinically stable. Magnetic resonance imaging (MRI) is the imaging test of choice to confirm diagnosis. Indications for empirical urgent corticosteroid therapy in patients with PA include hemodynamic instability, impaired consciousness, reduced visual acuity, and severe visual field defects. In patients with these severe neuro-ophthalmic signs, surgery should be considered. Surgery should preferably be performed within seven days of the onset of symptoms. Patients with mild and stable signs may be managed conservatively with careful monitoring. Treatment and long-term follow-up of patients with PA should be conducted by a multidisciplinary team consisting, amongst others, of an experienced pituitary neurosurgeon, an ophthalmologist, and an endocrinologist.
La apoplejía hipofisaria (AH) es un síndrome clínico agudo, potencialmente fatal, caracterizado por la aparición súbita de cefalea, vómitos, alteraciones visuales con afectación de los pares craneales y disminución del nivel de conciencia, provocado por la hemorragia y/o infarto de la glándula hipofisaria. Constituye uno de los cuadros clínicos de urgencia endocrinológica. Su diagnóstico requiere un índice elevado de sospecha y su manejo va a implicar a diferentes especialistas: médicos de urgencias, neurocirujanos, oftalmólogos y endocrinólogos. Sin embargo, no existe un consenso claro sobre cuál puede ser la mejor opción para su tratamiento.
Esta guía clínica no debe ser considerada como un estándar de práctica clínica. Solo pretende ofrecer una serie de recomendaciones para el diagnóstico y tratamiento de los pacientes con AH, basados en la evidencia médica disponible hasta la actualidad, que permitan armonizar en lo posible su manejo tanto en la fase aguda como en el seguimiento a largo plazo y que sirvan de ayuda a los profesionales implicados en el cuidado de estos pacientes. Sin embargo, el nivel de evidencia en el que se basan estas recomendaciones es bajo, al no existir estudios prospectivos y/o aleatorizados. Por tanto, sería necesaria la realización de este tipo de estudios para proporcionar un mayor nivel de evidencia, que permita optimizar el tratamiento de estos pacientes.
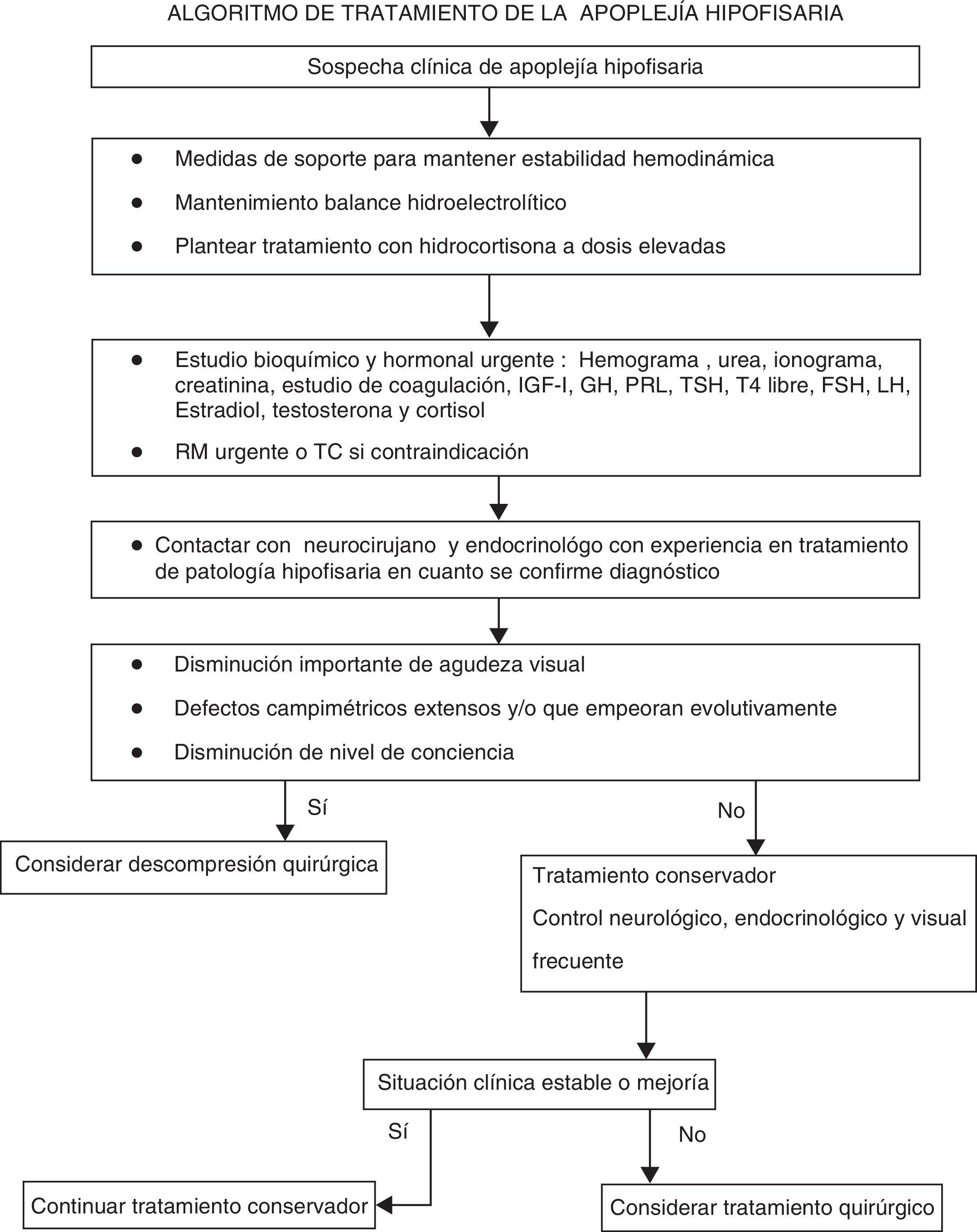
Resumen de recomedacionesRecomendaciones para la evaluación clínica inicialExamen clínico- •
El diagnóstico de apoplejía hipofisaria debe valorarse en aquellos pacientes con cefalea aguda grave, presenten o no síntomas neurooftalmológicos (✓).
- •
La evaluación clínica inicial siempre ha de recoger una anamnesis completa enfocada a detectar síntomas de disfunción hipofisaria así como una exploración física que incluya el examen de los pares craneales y una campimetría por confrontación (✓).
- •
Si la situación clínica del paciente lo permite, debe realizarse una campimetría o perimetría (analizador visual de Humphrey o el perímetro de Goldman) en las primeras 24h tras el inicio del cuadro (✓).
- •
A todos los pacientes en los que se sospeche una apoplejía hipofisaria se les han de extraer muestras de forma urgente para la medición de electrólitos, función renal, función hepática, coagulación, hemograma y función hipofisaria (cortisol, prolactina, tiroxina libre, TSH, IGF-1, GH, LH, FSH, estradiol en las mujeres en edad fértil y testosterona en los hombres) (IV, C).
- •
En todos los pacientes en los que se sospeche una apoplejía hipofisaria es necesario la realización de una resonancia magnética (RM) de forma urgente para confirmar el diagnóstico (III, B).
- •
Monitorización frecuente de balance hidroelectrolítico, medidas para mantener estabilidad hemodinámica y tratamiento con dosis altas de glucocorticoides (III, B).
- •
Las indicaciones para la administración urgente empírica de glucocorticoides en pacientes con AH son la inestabilidad hemodinámica, disminución de nivel de conciencia, disminución de la agudeza visual y defectos extensos en el campo visual (IV, C).
- •
Preferentemente se empleará hidrocortisona 100 a 200mg en bolo seguidos de 2-4mg por hora en infusión i.v. continua (✓).
- •
Una vez estabilizado el paciente, deberá ser trasladado a un centro donde exista disponibilidad de tratamiento neuroquirúrgico por vía transesfenoidal y posibilidad de supervisión estrecha desde el punto de vista oftalmológico y endocrinológico (✓).
- •
Los pacientes con alteración del nivel de conciencia y/o disminución de agudeza visual y/o defectos graves en el campo visual de presentación aguda, persistente o con deterioro progresivo, deberían ser tratados quirúrgicamente (III, B).
- •
La intervención debería realizarse preferentemente en los primeros 7 días tras la aparición de los síntomas (III, B).
- •
La intervención debería ser realizada por un neurocirujano con experiencia en cirugía transesfenoidal, de forma programada.
- •
Los pacientes sin sintomatología neurooftalmológica o con síntomas y/o signos leves y estables pueden ser manejados de forma conservadora, bajo supervisión estrecha (III, B).
- •
El control de sintomatología neurológica deberá realizarse inicialmente cada hora, posteriormente, si la evolución es favorable y el paciente está estable, se puede ampliar a 4 a 6h (✓).
- •
La agudeza visual y los defectos en el campo visual deben explorarse a diario hasta que se objetive una clara tendencia a la mejoría (✓).
- •
La función renal y las concentraciones de electrolíticos se controlarán cada 24 h o con más frecuencia si es necesario (✓).
En los pacientes con deterioro de la agudeza visual, del nivel de conciencia o empeoramiento de los defectos del campo visual deberá realizarse una RM urgente para planificar la descompresión quirúrgica, incluida la realización de derivación ventricular en caso de hidrocefalia (IV, C).
Recomendaciones para el seguimiento clínico en el postoperatorio inmediatoExamen clínico- •
Durante las primeras 24 o 48h tras la intervención, el paciente debe ser vigilado de forma muy estrecha para detectar las posibles complicaciones como la diabetes insípida, la pérdida de visión, la fístula de líquido cefalorraquídeo o el déficit de hormona adrenocorticotropa/cortisol. El balance hídrico ha de registrarse cada hora (✓).
- •
La agudeza visual, los movimientos oculares y la campimetría por confrontación deben explorarse periódicamente durante las primeras 48 h junto a la cama del paciente y cuando sea posible debe realizarse un examen instrumental (analizador Humphrey o perímetro de Goldman) (IV, C).
- •
En las primeras 24 o 48h deben evaluarse la función renal y los electrólitos plasmáticos y la osmolalidad plasmática y urinaria en caso de sospecharse una diabetes insípida. Estas determinaciones han de realizarse al menos una vez al día y si la situación clínica lo requiere deben llevarse a cabo con más frecuencia (IV, C).
- •
Se ha de medir el cortisol a las 09:00 h de la mañana al segundo o tercer día después de la intervención en pacientes sin evidencia previa de deficiencia de ACTH/cortisol. Para ello, será necesario interrumpir la administración de hidrocortisona la tarde de antes de la extracción (IV, C).
- •
En aquellos pacientes con déficit de ACTH/cortisol previo a la cirugía, el tratamiento con hidrocortisona debe continuarse hasta alcanzar la dosis de mantenimiento. Estos casos han de ser reevaluados a las 4 u 8 semanas para establecer si precisarán tratamiento indefinido (IV, C).
- •
La tiroxina libre y la TSH deben determinarse al tercer o cuarto día tras la cirugía. Si fueran normales deberían volver a medirse a las 4 u 8 semanas (IV, C).
- •
Si el paciente presentara un deterioro de la visión debe realizarse una RM de forma urgente y ser reevaluado por el equipo neuroquirúrgico sin demora (✓).
- •
Todos los pacientes que hayan sufrido una AH deben ser evaluados a las 4 u 8 semanas del episodio agudo. Debe realizarse un estudio de la función hipofisaria (determinaciones basales y, cuando estén indicadas, las pruebas de estímulo/supresión pertinentes), una exploración de los pares craneales así como una campimetría y de forma opcional una tomografía de coherencia óptica (TCO) (✓).
- •
Todos los pacientes que hayan sufrido una apoplejía deben ser evaluados anualmente mediante un estudio bioquímico y un estudio de la función hipofisaria que ha de incluir cortisol, tiroxina libre, TSH, LH, FSH, testosterona en varones, estradiol en mujeres en edad fértil, prolactina IGF-1 y tests dinámicos de cortisol y GH si clínicamente estuvieran indicados (✓).
- •
Aquellos pacientes que hayan sufrido una apoplejía y que presenten tumor residual precisarán seguimiento radiológico (RM) y, en aquellos casos en los que esté indicado, completarán el tratamiento con una nueva intervención quirúrgica, tratamiento médico o radioterapia (III, B).
- •
Se recomienda la realización de un control de RM a los 3 o 6 meses de la apoplejía. Si se detecta tumor residual o recurrencia se recomienda control anual durante los primeros 3 o 5 años y posteriormente cada 2 o 3 años (IV, C).
- •
Todos los pacientes requieren al menos una revisión anual; es recomendable que los pacientes sean seguidos por un equipo multidisciplinario con experiencia en enfermedades hipofisarias (endocrinólogos, neurocirujanos, oftalmólogos y radiólogos) (✓).
La presente guía se ha elaborado a raíz de la propuesta del comité científico del Grupo de Neuroendocrinología (GNE) de la Sociedad Española de Endocrinología y Nutrición (SEEN), dentro del programa de actualización de las guías de práctica clínica en neuroendocrinología. En el año 2006, el Grupo de Trabajo de Neuroendocrinología de la SEEN publicó la Guía clínica del diagnóstico y tratamiento de la apoplejía hipofisaria1. Tomando como base esta primera publicación, así como la guía británica de práctica clínica publicada en 20112, se ha trabajado en su actualización y adaptación al formato utilizado en la mayoría de las revistas médicas internacionales. Para ello, tras la revisión bibliográfica, se ha evaluado la calidad de la evidencia y el peso de las recomendaciones de acuerdo con el sistema propuesto por la Agency for Health Care Policy and Research (AHCPR), cuya denominación actual es Agency for Healthcare Research and Quality (AHRQ), en el año 20043.
Niveles de evidencia y grados de recomendación utilizados (basados en el sistema propuesto por la Agency for Health Care Policy and Research)Niveles de evidenciaIa: Evidencia obtenida de metanálisis de ensayos clínicos de alta calidad (controlados y aleatorizados).
Ib: Evidencia obtenida de al menos un ensayo clínico de alta calidad.
IIa: Evidencia obtenida de un estudio bien diseñado controlado sin aleatorización.
IIb: Evidencia obtenida de al menos un estudio bien diseñado casi experimental.
III: Evidencia obtenida de estudios descriptivos bien diseñados y de estudios de casos y controles.
IV: Opinión de expertos.
- A.
Nivel de evidencia ia o ib.
- B.
Nivel de evidencia iia, iib o iii.
- C.
Nivel de evidencia iv.
√Buena práctica clínica.
Tras la revisión bibliográfica de cada uno de los diferentes apartados, se presentan una serie de recomendaciones. Tras cada recomendación, aparece entre paréntesis el nivel de evidencia seguido del grado de recomendación.
DefiniciónEl término AH, inicialmente acuñado por Brougham et al. en 19504, se utiliza de forma clásica para referirse al síndrome clínico agudo, potencialmente fatal, caracterizado por la aparición súbita de cefalea, vómitos, alteraciones visuales y disminución del nivel de conciencia provocado por la hemorragia o infarto de la glándula hipofisaria. La primera descripción del síndrome fue publicada por Bailey5 en 1898. Se trata de una definición más clínica que patológica, ya que la hemorragia y/o el infarto hipofisario asintomáticos pueden ser hallazgos radiológicos, quirúrgicos o histopatológicos, y no se deberían diagnosticar como AH6,7.
La AH se puede presentar clínicamente de 2 formas:
- -
Aguda. Se considera una urgencia neuroendocrinológica, potencialmente mortal, que precisa descompresión neuroquirúrgica precoz en la gran mayoría de los casos6,7.
- -
Subaguda o subclínica. De evolución más lenta y larvada, con manifestaciones clínicas más leves, cuya incidencia sería más frecuente que la de la forma clásica y que podría ser tratada de forma conservadora al menos inicialmente7–9.
La incidencia de AH aguda en adenomas hipofisarios oscila entre un 0,6 y un 9% según las series analizadas8,10–13. En el caso de la forma subaguda o subclínica, dicha incidencia sube hasta un 14-22%10,12–14. Afecta más a pacientes entre la quinta y la sexta década de la vida y es algo más frecuente en varones (1,6/1)11–15.
Hasta en el 80% de los pacientes la apoplejía es la primera manifestación de un adenoma hipofisario7. Puede producirse tanto en adenomas hipofisarios funcionantes como no funcionantes11, aunque es más frecuente que aparezca en estos últimos, debido a la ausencia de un síndrome clínico de hiperfunción hormonal que alerte sobre la existencia del tumor. Se ha descrito su aparición en el seno de lesiones no adenomatosas, como craneofaringiomas, quistes hipofisarios e hipofisitis8.
El estudio anatomopatológico en los casos de AH muestra la presencia de infarto hemorrágico, hemorragia, necrosis o cambios quísticos intratumorales13,16. La mayoría de las series no hacen una distinción clara entre infarto puro y hemorragia o infarto hemorrágico. Algunos autores han encontrado que la hemorragia y el infarto hemorrágico se asocian más frecuentemente con la presencia de un factor precipitante, con un cuadro clínico más grave y con un pronóstico peor que el infarto no hemorrágico7. Sin embargo, el hallazgo de pequeñas áreas de hemorragia, infarto hemorrágico o infarto en las pruebas de imagen, durante la cirugía o en los estudios histopatológicos, se describe hasta en un 25% de los macroadenomas hipofisarios6,15,16, sin que en la mayoría de los casos se hayan producido manifestaciones clínicas evidentes, por lo que en estos casos no se pueden considerar como AH.
FisiopatologíaLas manifestaciones clínicas en la AH están causadas por un rápido aumento en el tamaño del contenido intraselar y el consiguiente incremento de la presión intraselar (47mm Hg de mediana en la AH frente a los 7-15mm Hg en condiciones normales), que produce una compresión mecánica de las vías ópticas y de las estructuras internas del seno cavernoso17. La hemorragia suele encapsularse dentro del tumor, pero con frecuencia se produce extravasación de sangre al espacio subaracnoideo, dando lugar a síntomas de irritación meníngea. En los grandes macroadenomas con extensión supraselar se puede producir una hidrocefalia obstructiva como complicación de una AH.
La compresión lateral puede afectar el contenido del seno cavernoso causando la parálisis de nervios oculomotores (oftalmoplejía) en el 70% de los pacientes, siendo el tercer par el más frecuentemente afectado18. La compresión superior sobre el quiasma óptico produce un deterioro de la agudeza visual y defectos campimétricos como la hemianopsia bitemporal en cerca del 75% de los pacientes8,10,12. La compresión inferior puede dar lugar a destrucción del suelo selar con fístula de líquido cefalorraquídeo17.
El aumento rápido y grave de la presión intraselar también va a ser responsable de la necrosis isquémica de la hipófisis anterior, por compresión directa de la hipófisis normal y disminución del aporte sanguíneo a la glándula17. Como consecuencia de dicha destrucción, se va a producir un cuadro de hipopituitarismo19,20. Será necesaria una destrucción glandular del 75-90% para que se produzcan déficit hormonales permanentes17.
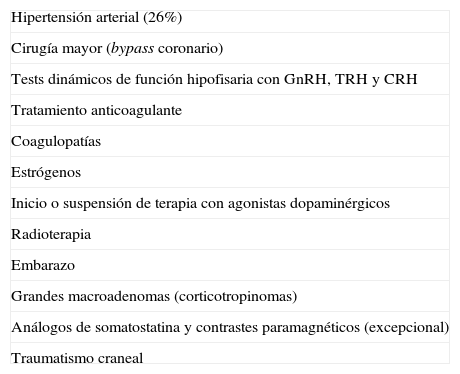
Factores precipitantesHasta en el 40% de los casos de AH se han podido identificar factores precipitantes12 (tabla 1). Los más frecuentes son la hipertensión arterial (26%)8,12, la anticoagulación y la cirugía mayor, especialmente la cirugía de bypass coronario21,22. En esta última, debido sobre todo a las fluctuaciones de presión arterial y al uso de terapia anticoagulante23.
Factores precipitantes en la apoplejía hipofisaria
| Hipertensión arterial (26%) |
| Cirugía mayor (bypass coronario) |
| Tests dinámicos de función hipofisaria con GnRH, TRH y CRH |
| Tratamiento anticoagulante |
| Coagulopatías |
| Estrógenos |
| Inicio o suspensión de terapia con agonistas dopaminérgicos |
| Radioterapia |
| Embarazo |
| Grandes macroadenomas (corticotropinomas) |
| Análogos de somatostatina y contrastes paramagnéticos (excepcional) |
| Traumatismo craneal |
Otros factores favorecedores o precipitantes incluyen los tests dinámicos utilizados para evaluar la función hipofisaria, como los de GnRH, TRH, CRH e hipoglucemia insulínica. La apoplejía se presentó dentro de las 2 h posteriores a los tests en el 83% de los pacientes, y dentro de las 88 h posteriores en todos los casos24.
La anticoagulación, las coagulopatías, el inicio o suspensión de tratamiento con agonistas dopaminérgicos10 o estrógenos, el tratamiento con radioterapia16, el embarazo25 y los traumatismos craneales también pueden inducir el desarrollo de una AH. La resección parcial de un macroadenoma constituye otro factor de riesgo debido al compromiso de la irrigación en el remanente posquirúrgico26.
Se indica que los adenomas productores de GH y de ACTH, así como los grandes adenomas no funcionantes, especialmente los corticotropinomas silentes, tienen un riesgo elevado de presentar una apoplejía27 (tabla 1).
ClínicaEl cuadro clínico suele evolucionar en el curso de horas o días7. Va a depender de la agudeza y del volumen de la hemorragia, de la presencia de manifestaciones endocrinológicas asociadas y de las estructuras paraselares afectadas. Su aparición de forma aguda con graves déficit neurológicos evoluciona en 24 o 48h y se considera una urgencia neuroendocrinológica potencialmente mortal, que va a requerir una descompresión quirúrgica inmediata7,8. En la forma subaguda o «apoplejía hipofisaria subclínica», la evolución de la hemorragia y/o infarto hipofisario puede ser más lenta e insidiosa, con manifestaciones clínicas leves7–9.
El síntoma más frecuente es la cefalea, que suele ser de localización retroorbitaria, aunque también puede describirse como bifrontal o suboccipital. Su instauración es brusca e intensa, se acompaña con frecuencia de vómitos y suele preceder a los demás síntomas12,13,17,28.
La afectación visual es frecuente y se manifiesta como paresia oculomotora. En el 50% de los pacientes se detectan síntomas de afectación del iii par10. La diplopía aparece en un número elevado de casos (67%)8. También puede producirse disminución de agudeza visual de grado variable (desde defectos campimétricos a amaurosis bilateral) y deterioro del nivel de conciencia (fig. 1).

Excepcionalmente se han descrito crisis epilépticas, hemiplejía por compresión del tronco cerebral y diabetes insípida4,10 (tabla 2).
Síntomas y signos que aparecen en la apoplejía hipofisaria
| Síntomas y signos | Porcentaje |
| Cefalea | 95 |
| Náuseas | 80 |
| Reducción del campo visual | 71 |
| Afección de pares craneales | |
| iii | 67 |
| iv | 4 |
| vi | 29 |
| Reducción de la agudeza visual | 66 |
| Vómitos | 57 |
| Fotofobia | 49 |
| Pirexia | 20 |
| Disminución del nivel de conciencia | 11 |
En cuanto a la síntomatología debida a disfunción hormonal, casi el 80% de los pacientes presenta alguna deficiencia de las hormonas de la hipófisis anterior al inicio del cuadro, en relación con la existencia de un macroadenoma no diagnosticado29. El déficit de ACTH/cortisol está presente en el 70% de los pacientes. Es el más importante por el riesgo que implica, puesto que la insuficiencia suprarrenal aguda es la causa principal de la mortalidad y se manifiesta en forma de inestabilidad hemodinámica. El déficit de GH, LH/FSH y el de TSH suelen aparecer en porcentajes superiores al 80, 75 y 60%, respectivamente11,12,18,19.
Se ha descrito la presencia de hiponatremia en el 40% de los pacientes, bien en relación con secreción inadecuada de hormona antidiurética o con hipocortisolismo8. La aparición de diabetes insípida se produce con menor frecuencia21.
Las concentraciones de prolactina pueden variar dependiendo de la extensión de la necrosis isquémica hipofisaria. Algunos autores han sugerido que concentraciones normales o discretamente elevadas indicarían un grado menor de destrucción glandular y, por tanto, mayor probabilidad de recuperación de la función hipofisaria tras cirugía descompresora17,19.
DiagnósticoLa forma de instauración aguda de la apoplejía hipofisaria remeda a menudo el cuadro clínico de otras urgencias neurológicas más frecuentes, por lo que puede ser difícil establecer inicialmente el diagnóstico de esta entidad. Deberemos realizar el diagnóstico diferencial principalmente con la hemorragia subaracnoidea y la meningitis bacteriana. El infarto del mesencéfalo (oclusión de la arteria basilar) o la trombosis del seno cavernoso, aunque menos frecuentes, también deben ser tenidos en cuenta.
La presencia simultánea o sucesiva de los siguientes síntomas y signos va a indicar de forma consistente la existencia de una apoplejía hipofisaria:
- 1.
Cefalea continua e intensa de aparición brusca y predominio retrorbitario.
- 2.
Diplopía asociada a reducción del campo visual y/o de la agudeza visual.
- 3.
Alteración del nivel de conciencia.
La RM es la prueba radiológica de elección, ya que confirma la sospecha diagnóstica en más del 90% de los casos y nos va a permitir realizar el diagnóstico diferencial con otras urgencias neurológicas. En caso de deterioro visual debe realizarse sin demora. La tomografía computarizada (TC) solo diagnostica menos del 30% de las apoplejías, aunque objetiva la presencia de una masa selar en más del 80%11–14. La RM suele mostrar en la fase inicial una masa hipofisaria con señal heterogénea hiperintensa predominantemente en las imágenes potenciadas en T1 e hipointensa en las imágenes potenciadas en T2. En las imágenes de secuencias de eco de gradiente en T2 se observa la hemorragia intratumoral en fase aguda como una lesión muy oscura que contrasta con las estructuras colindantes. En las apoplejías subagudas, la hemorragia se ve en las imágenes de secuencia de eco de gradiente en T2 como un anillo oscuro30.
La prueba de elección para evaluar la posible lesión de vía óptica es la campimetría o perimetría. Aunque la perimetría visual (analizador visual de Humphrey o el perímetro de Goldman) permite conocer de forma objetiva el grado de afectación del campo visual, no es útil para establecer un pronóstico acerca de la evolución del daño campimétrico. La TCO es una exploración sencilla que obtiene una imagen de la retina y del disco óptico y cuantifica la pérdida neuronal a este nivel. La TCO es de gran utilidad para el estudio de la enfermedad macular y cada vez tiene más aplicaciones para valorar la pérdida neuronal en el sistema nervioso central, extrapolando lo ocurrido en la capa de fibras nerviosas de la retina (CFNR) a nivel macular y peripapilar. Esta prueba podría ser útil para el diagnóstico y el seguimiento de los pacientes con compresión quiasmática por tumores hipofisarios. Diferentes estudios han comunicado que un adelgazamiento de la CFNR a los 3 meses de la descompresión quirúrgica se asocia a un mal pronóstico27,31,32, aunque un trabajo concluye que no siempre los hallazgos de la TCO se correlacionan con las alteraciones campimétricas33. Así pues, se necesitan estudios prospectivos para determinar el valor pronóstico de la TCO como predictora de la recuperación visual tras el tratamiento de los pacientes con adenomas hipofisarios.
Recomendaciones para la evaluación clínica en la presentación del cuadroExamen clínico- •
El diagnóstico de apoplejía hipofisaria debe valorarse en aquellos pacientes con cefalea severa aguda, presenten o no síntomas neurooftalmológicos (✓).
- •
Los pacientes que han sido diagnosticados de un adenoma hipofisario deben ser advertidos de la posibilidad de apoplejía y de sus síntomas, sobre todo aquellos casos que presentan factores precipitantes (tratamiento anticoagulante, cirugía con circulación extracorpórea, cirugía mayor, etc.) (✓).
- •
La evaluación clínica inicial siempre ha de recoger una anamnesis completa enfocada a detectar síntomas de disfunción hipofisaria, así como una exploración física que incluya el examen de los pares craneales y una campimetría por confrontación (✓).
- •
Si la situación clínica del paciente lo permite, debe realizarse una campimetría o perimetría (analizador visual de Humphrey o el perímetro de Goldman) en las primeras 24h tras el inicio del cuadro (✓).
- •
A todos los pacientes en los que se sospeche una apoplejía hipofisaria se les han de extraer muestras de forma urgente para la medición de electrólitos, función renal, función hepática, coagulación, hemograma y función hipofisaria (cortisol, prolactina, tiroxina libre, TSH, IGF-1, GH, LH, FSH, estradiol en las mujeres en edad fértil y testosterona en los hombres) (IV, C).
- •
En pacientes hemodinámicamente inestables en los que es preciso iniciar las medidas de soporte sin demora, antes de administrar la hidrocortisona i.v., se deben de tomar muestras de sangre para la determinación de hormonas tiroideas (TSH, tiroxina libre) y cortisol (IV, C).
- •
Si en el momento en el que el paciente es atendido por primera vez no fuera posible enviar directamente las muestras al laboratorio se ha de guardar una muestra de suero para su análisis posterior (✓).
- •
En todos los pacientes en los que se sospeche una apoplejía hipofisaria es necesario la realización de una RM de forma urgente para confirmar el diagnóstico (III, B).
- •
La TC está indicada si la RM está contraindicada o no es posible realizarla (IV, C).
El tratamiento de la AH se basa en 2 pilares terapéuticos:
Tratamiento médico- -
Se va a basar en el uso de glucocorticoides en dosis altas, el control de las alteraciones hidroelectrolíticas, el mantenimiento de la estabilidad hemodinámica si es preciso y el tratamiento de los déficit hormonales8,12,15.
- -
El uso de glucocorticoides en dosis elevadas se basa en la presencia de insuficiencia suprarrenal y en su efecto antiinflamatorio y antiedematoso. La hipocortisolemia produce una menor respuesta vascular a las catecolaminas, lo que favorece la inestabilidad hemodinámica. También aumenta la liberación de vasopresina, lo que da lugar a la disminución de la excreción de agua libre, con aparición de hiponatremia. Por tanto, será necesario instaurar un tratamiento con glucocorticoides en pacientes con inestabilidad hemodinámica y/o síntomas y signos de hipocortisolismo antes de conocer los resultados de las concentraciones de cortisol2,34.
- -
Las guías clínicas publicadas en 20112 abogan por la utilización de hidrocortisona, en bolo inicial de 100 a 200mg i.v., seguido de perfusión continua (2-4mg/h) o 50 a 100mg por vía intramuscular (i.m.) cada 8 o 6 h. La razón para la indicación de esta pauta es que la administración de hidrocortisona en bolos por vía i.v. cada 6 u 8 h da lugar a saturación de la proteína transportadora de cortisol, con lo que gran parte de la hidrocortisona se filtrará a nivel renal sin llegar a realizar su efecto. Los esteroides potentes de vida media larga (dexametasona, 2 a 16mg al día) no se consideran una buena opción, aunque pueden ser útiles inicialmente para disminuir el edema si no se realiza descompresión quirúrgica precoz. Puede añadirse manitol, según las necesidades.
- -
Si hubiese mejoría tras el episodio agudo, se recomienda disminuir la dosis de glucocorticoides rápidamente hasta 20 a 30mg de hidrocortisona al día por vía oral divididos en 3 dosis2,34.
- •
Monitorización frecuente de balance hidroelectrolítico, medidas para mantener estabilidad hemodinámica y tratamiento con dosis altas de corticoides (III, B).
- •
Las indicaciones para la administración urgente empírica de glucocorticoides en pacientes con AH son la inestabilidad hemodinámica, disminución de nivel de conciencia, disminución de la agudeza visual y defectos extensos en el campo visual (IV, C).
- •
Preferentemente se empleará hidrocortisona 100 a 200mg en bolo seguidos de 2-4mg por hora en infusión i.v. continua. Alternativamente se pueden administrar de 50 a 100mg por vía i.m. cada 6h. Previamente se obtendrán muestras sanguíneas para determinar concentraciones hormonales y de analítica general como se indica en el apartado pruebas de laboratorio. La dexametasona en dosis elevadas puede utilizarse inicialmente en pacientes con edema cerebral (IV, C).
- •
En los pacientes que no cumplan los criterios anteriores, pero con concentraciones de cortisol a las 09:00 h menores de 550nmol/l (15μg/dl), también se administrarán esteroides con la misma pauta (IV, C).
- •
Una vez estabilizado el paciente, deberá ser trasladado a un centro donde exista disponibilidad de tratamiento neuroquirúrgico por vía transesfenoidal y posibilidad de supervisión estrecha desde el punto de vista oftalmológico y endocrinológico (✓).
Será necesaria una descompresión quirúrgica, preferentemente por vía transesfenoidal. Sin embargo, no hay acuerdo sobre si siempre se debe realizar tratamiento quirúrgico ni sobre el momento en que es más conveniente realizarlo8,12,18. No hay estudios clínicos prospectivos y aleatorizados en los que basar las recomendaciones y la evidencia científica disponible hasta la actualidad proviene de estudios retrospectivos y casos clínicos. El abordaje por vía transesfenoidal se prefiere en la mayoría de los pacientes debido a su baja morbimortalidad.
Se insiste en la necesidad de que, una vez estabilizado, el paciente sea tratado en centros con disponibilidad de tratamiento neuroquirúrgico, mejor con experiencia en cirugía transesfenoidal2,34, y posibilidad de supervisión estrecha desde el punto de vista oftalmológico y endocrinológico. En la guía clínica británica para el manejo de la AH se considera cirujano experto aquel que realiza 5 o más procedimientos por vía transesfenoidal al año2.
En la mayoría de las series publicadas se recomienda tratamiento quirúrgico urgente cuando exista disminución de nivel de conciencia, afectación hipotalámica, amaurosis de aparición brusca o disminución de la agudeza visual8,12,34,35. También se recomienda en casos de empeoramiento de los defectos campimétricos y/o deterioro progresivo de la agudeza visual y/o de la sintomatología neurológica8,12.
En las series retrospectivas más antiguas se aboga por la realización en todos los casos de un tratamiento quirúrgico descompresivo8, basándose en la mejoría de la agudeza visual, de los defectos campimétricos, de la oftalmoplejía y de los déficit hormonales observada en la mayoría de los pacientes28. Esta mejoría se produce ya en el postoperatorio inmediato y continúa varias semanas después de la cirugía8. En diferentes trabajos se ha constatado que la mejoría es más completa si se realiza la intervención en los primeros 7 días desde el inicio de los síntomas8,13,36,37.
Recomendaciones para el tratamiento quirúrgico- •
Los pacientes con alteración del nivel de conciencia y/o disminución de agudeza visual y/o defectos graves en el campo visual de presentación aguda, persistente o con deterioro progresivo, deberían ser tratados quirúrgicamente (III, B).
- •
La intervención debería realizarse preferentemente en los primeros 7 días tras la aparición de los síntomas (III, B).
- •
La intervención debería ser realizada por un neurocirujano con experiencia en cirugía transesfenoidal, de forma programada. La descompresión urgente por el neurocirujano de guardia sin experiencia en esta cirugía debería reservarse solo para los casos que por su gravedad requieran intervención inmediata (IV, C).
En estudios retrospectivos más recientes6,38–40 se señala que los pacientes con afectación preferentemente oculomotora, sin disminución del nivel de conciencia y con mejoría visual en los primeros días, podrían ser tratados exclusivamente de forma conservadora. En estos casos, el pronóstico desde el punto de vista oftalmológico y endocrinológico no sería diferente del de los pacientes tratados quirúrgicamente. Sin embargo, hay que tener en cuenta el sesgo que supone el que los pacientes tratados con cirugía solían ser aquellos que presentaban síntomas graves al inicio o evolucionaron peor tras realizar tratamiento conservador.
Otro subgrupo de pacientes susceptibles de tratamiento conservador, sería el de aquellos con presentación clínica larvada (apoplejía hipofisaria subclínica)14,40.
Esta situación es aplicable especialmente a pacientes con adenomas secretores de prolactina. En estos casos, el tratamiento con un agonista dopaminérgico no sólo controlará las concentraciones de prolactina, sino que también reducirá el tamaño tumoral35.
Se ha intentado buscar diferentes criterios para tomar decisiones respecto del tipo de tratamiento inicial. Se debe considerar la forma de presentación de la AH, la evolución clínica, una buena respuesta al tratamiento glucocorticoideo y la disponibilidad de un neurocirujano experto36. Algunos trabajos han propuesto el análisis de las características radiológicas38,41. En estudios no controlados se ha comunicado que la presencia de una sola área hipodensa en el seno del tumor, así como una involución temprana de la lesión, se asocian con más frecuencia a una resolución espontánea del cuadro, lo que abogaría por el mantenimiento del tratamiento conservador en estos casos38.
Un trabajo recientemente publicado propone utilizar una escala de puntuación que permite establecer grados de gravedad de la AH, basada en la escala de Glasgow, agudeza visual, presencia de defectos en el campo visual y grado de parálisis oculomotora42. Esta escala podría constituir una herramienta para la valoración de los pacientes basada en datos objetivos.
Recomendaciones para el tratamiento conservador- •
Los pacientes sin sintomatología neurooftalmológica o con síntomas y/o signos leves y estables pueden ser manejados de forma conservadora, bajo supervisión estrecha (III, B).
- •
El control de sintomatología neurológica deberá realizarse inicialmente cada hora, posteriormente, si la evolución es favorable y el paciente está estable, se puede ampliar a 4 a 6h (✓).
- •
La agudeza visual y los defectos en el campo visual deben explorarse a diario hasta que se objetive una clara tendencia a la mejoría (✓).
- •
La función renal y los niveles electrolíticos se controlarán cada 24h o con más frecuencia si es necesario (✓).
- •
En los pacientes con deterioro de la agudeza visual, del nivel de conciencia o empeoramiento de los defectos del campo visual, deberán realizarse una RM urgente para planificar la descompresión quirúrgica, incluida la realización de derivación ventricular en caso de hidrocefalia (IV, C).
- •
La decisión de realizar tratamiento quirúrgico o manejar de forma conservadora al paciente con AH debería ser tomada por un equipo multidisciplinario que incluya un neurocirujano, un endocrinólogo y un oftalmólogo (✓).
- •
Las posibilidades de tratamiento y la decisión sobre el mismo deberían explicarse con claridad al paciente y, si es posible, debería obtenerse un consentimiento informado (✓).
- •
Debería proporcionarse a los pacientes o a sus familiares una hoja explicativa con información sencilla y clara sobre los tumores hipofisarios y la AH (✓).
Los cuidados durante el postoperatorio de los pacientes intervenidos por una AH son similares a los aplicados en la cirugía de los tumores hipofisarios.
La diabetes insípida posquirúrgica aparece hasta en el 16% de los pacientes con apoplejía hipofisaria intervenidos8. Otras posibles complicaciones postoperatorias son: el déficit de cortisol, la pérdida de visión, la fístula de líquido cefalorraquídeo y la meningitis.
La función suprarrenal y tiroidea deben ser evaluadas en el postoperatorio inmediato. El cortisol extraído a las 9 de la mañana es la prueba de elección para la evaluación inicial del paciente recientemente intervenido43. La función tiroidea (TSH y tiroxina libre) debe de ser medida al tercer o cuarto día tras la cirugía y, si fuera normal, ha de ser evaluada de nuevo a las 6/8 semanas.
La agudeza visual, los defectos campimétricos y la parálisis oculomotora mejoran en la mayoría de los pacientes después de la descompresión quirúrgica8,10,11. Tal mejoría se observa en el postoperatorio inmediato y a menudo continúa semanas después de la cirugía44.
Recomendaciones para el seguimiento clínico en el postoperatorio inmediatoExamen clínico- •
Durante las primeras 24 o 48h tras la intervención, el paciente debe ser vigilado de forma muy estrecha para detectar las posibles complicaciones como la diabetes insípida, la pérdida de visión, la fístula de líquido cefalorraquídeo o el déficit de ACTH/cortisol. El balance hídrico ha de registrarse cada hora (✓).
- •
La agudeza visual, los movimientos oculares y la campimetría por confrontación deben explorarse periódicamente durante las primeras 48h junto a la cama del paciente y cuando sea posible debe realizarse un examen instrumental (analizador Humphrey o perímetro de Goldman) (IV, C).
- •
En las primeras 24 o 48h deben evaluarse la función renal y los electrólitos plasmáticos y la osmolalidad plasmática y urinaria en caso de sospecharse una diabetes insípida. Estas determinaciones han de realizarse al menos una vez al día y si la situación clínica lo requiere deben llevarse a cabo con más frecuencia (IV, C).
- •
Se ha de medir el cortisol a las 09:00 h de la mañana al segundo o tercer día después de la intervención en pacientes sin evidencia previa de deficiencia de ACTH/cortisol. Para ello, será necesario interrumpir la administración de hidrocortisona la tarde de antes de la extracción (IV, C).
- •
En aquellos pacientes con déficit de ACTH/cortisol previo a la cirugía, el tratamiento con hidrocortisona debe continuarse hasta alcanzar la dosis de mantenimiento. Estos casos han de ser reevaluados a las 4 u 8 semanas para establecer si precisarán tratamiento indefinido (IV, C).
- •
La tiroxina libre y la TSH deben determinarse al tercer o cuarto día tras la cirugía. Si fueran normales deberían volver a medirse a las 4 u 8 semanas (IV, C).
- •
Si el paciente presentara un deterioro de la visión debe realizarse una RM de forma urgente y ser reevaluado por el equipo neuroquirúrgico sin demora (✓).
Diferentes estudios han demostrado que la recuperación funcional de la hipófisis anterior se produce de forma parcial o total en un 50% de los pacientes que han sufrido una AH17.
Tras la AH se va a producir una evolución durante los siguientes días o meses a una situación de hipopituitarismo parcial o completo, que puede ser transitorio o permanente. Cerca del 80% de los pacientes necesitará algún tipo de tratamiento hormonal sustitutivo. El déficit de GH es el más frecuente (80-90%), seguido por el déficit de gonadotropinas (60-80%) y el de ACTH/cortisol (60-80%), el de TSH (50-60%) y el de arginina vasopresina (10-25%)8,18,19,38.
Con frecuencia, la mejoría de las manifestaciones visuales tras el tratamiento inicial se mantiene e incluso continúa a lo largo del seguimiento. La recuperación va a ser más improbable en pacientes que han presentado pérdida de visión uni o bilateral38. El síntoma que primero se recupera es la diplopía, mientras que la reducción del campo visual mejora más lentamente30,44. La TCO aporta una información inédita hasta ahora en la enfermedad hipofisaria, es una técnica fácil de realizar y no presenta contraindicaciones, por lo que se debería incluir en la exploración rutinaria de estos pacientes en el seguimiento a medio plazo, ya que puede ayudar a conocer el pronóstico de las lesiones del campo visual.
La recurrencia de la apoplejía, así como el crecimiento o recidiva tumoral, se han comunicado tanto en pacientes tratados de forma conservadora como en pacientes tratados con cirugía4,9,18,39. Por lo tanto, todos los pacientes deberán ser vigilados de forma prolongada para detectar posibles recurrencias45.
Recomendaciones para el seguimiento a medio y largo plazo- •
Todos los pacientes que hayan sufrido una AH deben ser evaluados a las 4/8 semanas del episodio agudo. Debe realizarse un estudio de la función hipofisaria (determinaciones basales y cuando estén indicadas, las pruebas de estímulo/supresión pertinentes), una exploración de los pares craneales así como una campimetría y de forma opcional una TCO (✓).
- •
Todos los pacientes que hayan sufrido una apoplejía deben ser evaluados anualmente mediante un estudio bioquímico y un estudio de la función hipofisaria que ha de incluir cortisol, tiroxina libre, TSH, LH, FSH, testosterona en varones, estradiol en mujeres en edad fértil, prolactina IGF-1 y tests dinámicos de cortisol y GH si clínicamente estuvieran indicados (✓).
- •
Aquellos pacientes que hayan sufrido una apoplejía y que presenten tumor residual precisarán seguimiento radiológico (RM) y, en aquellos casos en los que esté indicado, completarán el tratamiento con una nueva intervención quirúrgica, tratamiento médico o radioterapia (III, B).
- •
Se recomienda la realización de un control de RM a los 3 o 6 meses de la apoplejía. Si se detecta tumor residual o recurrencia se recomienda control anual durante los primeros 3 o 5 años y posteriormente cada 2 o 3 años (IV, C).
- •
Todos los pacientes requieren al menos de una revisión anual, es recomendable que los pacientes sean seguidos por un equipo multidisciplinario con experiencia en enfermedad hipofisaria (endocrinólogos, neurocirujanos, radiólogos) (✓).
Es necesaria la realización de estudios prospectivos y/o controlados y/o aleatorizados que permitan responder las siguientes preguntas:
- •
¿En qué momento es mejor realizar la descompresión quirúrgica cuando es necesaria?
- •
¿Qué pacientes se van a beneficiar del tratamiento quirúrgico frente al tratamiento conservador? ¿Se puede desarrollar y validar una escala de gradación clínica que permita distinguir a estos pacientes?
- •
¿Cuál es el pronóstico a largo plazo desde el punto de vista neurooftalmológico y endocrinológico de los pacientes tratados quirúrgicamente frente a los que recibieron tratamiento conservador?
Los autores declaran no tener ningún conflicto de intereses.
Los nombres de los componentes del Grupo de Trabajo de Neuroendocrinología están relacionados en el anexo al final del artículo.
