


La hipercalcemia hipocalciúrica familiar, también denominada hipercalcemia benigna familiar, es una causa poco frecuente de hipercalcemia. Se debe a una mutación del receptor sensible al calcio que se hereda de forma autosómica dominante con alta penetrancia. En general, los pacientes no presentan síntomas y los casos heterocigotos se diagnostican en la infancia o en la edad adulta al estudiar una hipercalcemia detectada de forma incidental. Se caracteriza por hipercalcemia moderada, con paratirina normal o levemente elevada y calciuria baja. Es importante establecer el diagnóstico por su benignidad y porque es una situación que no requiere tratamiento quirúrgico, a diferencia del hiperparatiroidismo que precisa paratiroidectomía en el 50% de los casos. Presentamos 3 casos pertenecientes a la misma familia con hipercalcemia hipocalciúrica familiar y a continuación se realiza una revisión actualizada del tema.
Familial hypocalciuric hypercalcemia, also denominated familial benign hypercalcemia, is an uncommon cause of hypercalcemia. It is caused by mutations of the calcium-sensing receptor, which are inherited in an autosomal dominant high-penetrance fashion. Generally, patients are asymptomatic, and heterozygote cases are diagnosed in childhood or adulthood, when diagnostic work-up of an incidentally discovered hypercalcemia ensues. This disorder is characterized by moderate hypercalcemia, with normal parathormone levels and low urine calcium excretion. It is very important to diagnose this condition, as it does not require surgical procedures, unlike primary hyperparathyroidism, which needs parathyroidectomy in 50% of cases. We present 3 cases of familial hypocalciuric hypercalcemia belonging to the same family, and provide an updated review on the topic.
Las causas principales de hipercalcemia son hiperparatiroidismo y neoplasias, aunque siempre debemos considerar etiologías menos frecuentes, como intoxicación por vitamina D, enfermedades granulomatosas, tirotoxicosis, inmovilización prolongada e hipercalcemia hipocalciúrica familiar (HHF)1.
La HHF supone aproximadamente el 2% de todas las causas de hipercalcemia. Se debe a la mutación del gen que codifica el receptor sensible al calcio, de tal forma que hay una insensibilidad generalizada al ion calcio en las células tubulares renales y en las glándulas partiroideas, por lo que no se produce la esperada disminución de la liberación de paratirina (PTH) y el incremento de la eliminación renal de calcio ante sus concentraciones elevadas2. Su curso generalmente es benigno y no se relaciona con manifestaciones clínicas, aunque un pequeño porcentaje de pacientes pueden presentar pancreatitis de repetición o condrocalcinosis3.
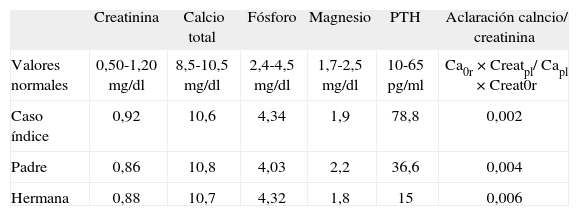
CASO CLÍNICOVarón de 15 años en que el estudio de ginecomastia peripuberal revela hipercalcemia. Sus antecedentes personales carecían de interés: nacido de embarazo y parto normales con posterior desarrollo psicomotor y pondostatural adecuados. Los datos analíticos se caracterizaban por hemograma dentro de la normalidad, bioquímica sin interés, salvo calcio plasmático de 10,8 (8,5-10,5) mg/dl; fósforo, 4,57 (2,5-4,5) mg/dl; fosfatasa alcalina, 109 (40–129) U/l; proteínas totales, 7,3 (6–8,5) g/dl, y albúmina, 4,5 (3,4-4,9) g/dl. El metabolismo fosfocálcico se reevaluó una vez finalizado el crecimiento y destacaba: calcio, 10,6mg/dl; calcio iónico, 4,11 (4–5,3) mg/dl; fósforo, 4,34; magnesio, 1,9 (1,7- 2,5) mg/dl; excreción de calcio, 77,9mg/24h; excreción de fosfato, 1.012mg/24h; fosfatasa alcalina, 88 (35–104) U/l; PTH, 78,8 (10–65) pg/ml; calcidiol, 33 (8–75) ng/ml. Interrogado específicamente, no refería clínica de hipercalcemia ni antecedentes familiares de alteraciones del metabolismo fosfocálcico y la ingesta de calcio era adecuada. La gammagrafía de paratiroides no mostró captaciones patológicas y permitió descartar adenoma e hiperplasia de las glándulas. Las mismas determinaciones analíticas se repitieron en 6 meses, en que se obtuvo resultados similares: hipercalcemia leve, hipocalciuria y PTH discretamente elevada. Ante la hipercalcemia y la hipocalciuria persistentes, se sospechó una hipercalcemia hipocalciúrica familiar. Con la finalidad de confirmar esta posibilidad se calculó el cociente aclaramiento de calcio/aclaramiento de creatinina, que fue 0,0029. Este valor, inferior a 0,01, se consideró indicativo de HHF. Con este diagnóstico se procedió a estudiar a sus familiares de primer grado: padre, madre y única hermana.
El padre, de 45 años de edad, sin antecedentes personales de interés, no presentaba clínica de hipercalcemia. Respecto al metabolismo fosfocálcico, se obtuvieron los siguientes resultados: calcio, 10,6mg/dl; calcio iónico, 4,23mg/dl; fósforo, 4,03mg/dl; magnesio, 2,2mg/dl; excreción de calcio, 58,9mg/24h; excreción de fosfato, 414,5mg/24h; PTH, 36,6pg/ml, y calcidiol, 13ng/ml. El cociente aclaramiento de calcio/aclaramiento de creatinina fue 0,004.
La hermana de 19 años de edad no refería antecedentes de interés ni síntomas de hipercalcemia. Los resultados analíticos son los citados a continuación: calcio, 10,7mg/dl; calcio iónico, 4,28mg/dl; fósforo, 4,32mg/dl; magnesio, 1,8mg/dl; excreción de calcio, 68,8mg/24h; excreción de fosfato, 386,5mg/24h; PTH, 15pg/ml, y vitamina D cacidiol, 28ng/dl. El cociente aclaramiento de calcio/aclaramiento de creatinina fue 0,006. La madre no mostraba alteraciones en el metabolismo fosfocálcico. Todos los resultados analíticos se resumen en la tabla 1.
Datos analíticos de los pacientes. Destaca la hipercalcemia concomitante con concentraciones normales de paratirina (PTH) y relativamente bajas de calcio en orina de 24h
| Creatinina | Calcio total | Fósforo | Magnesio | PTH | Aclaración calncio/ creatinina | |
| Valores normales | 0,50-1,20mg/dl | 8,5-10,5mg/dl | 2,4-4,5mg/dl | 1,7-2,5mg/dl | 10-65pg/ml | Ca0r×Creatpl/ Capl×Creat0r |
| Caso índice | 0,92 | 10,6 | 4,34 | 1,9 | 78,8 | 0,002 |
| Padre | 0,86 | 10,8 | 4,03 | 2,2 | 36,6 | 0,004 |
| Hermana | 0,88 | 10,7 | 4,32 | 1,8 | 15 | 0,006 |
La HHF es una causa poco común de hipercalcemia que se produce por una mutación inactivante del receptor sensible al calcio. Dicho receptor se expresa en múltiples tejidos: glándulas paratiroideas, riñón, médula ósea, osteoclastos, células C del tiroides, células secretoras de gastrina y en algunas áreas del cerebro4,5. Su principal misión es regular el balance del calcio modificando la función de las glándulas paratiroideas y el manejo de calcio en el riñón. En la HHF la mutación del receptor disminuye su sensibilidad al calcio, de tal forma que en las glándulas paratiroideas son necesarias concentraciones más elevadas de calcio para disminuir la liberación de PTH. En el riñón el defecto del receptor conlleva una mayor reabsorción de calcio y magnesio y la consecuencia es hipercalcemia, hipocalciuria absoluta (inferior a 5mg/kg/día) o relativa para el valor de calcio plasmático (cociente aclaramiento de calcio/aclaramiento de creatinina menor de 0,01) y frecuentemente hipermagnesemia6.
El grado de hipercalcemia parece relacionarse con el tipo de defecto genético7. La mutación de un único alelo del sensor de calcio (heterocigosis) genera el fenotipo característico de la HHF, previamente descrito. Las características clínicas de esta entidad presentan un continuo en el que la secreción de PTH y la reabsorción tubular de calcio se ven directamente condicionadas por la gravedad de la disfunción que la mutación comporta para el sensor de calcio8. Mientras que los pacientes con un solo alelo mutado habitualmente son diagnosticados de forma incidental o por cribado de familiares de afectados, las situaciones de homocigosis (2 alelos mutados) comportan un hiperparatiroidismo neonatal grave, con hipercalcemias mayores de 15mg/dl9,10. Este trastorno amenaza la vida del neonato y precisa de paratiroidectomía urgente. La correlación genotipo-fenotipo en los trastornos del sensor del calcio se estableció inicialmente en estudios realizados en ratas que carecían del receptor sensible al calcio (los heterocigotos presentan un síndrome similar a la HHF, mientras que los homocigotos presentan hiperparatiroidismo neonatal grave)3. Posteriormente, múltiples estudios de agregación familiar en humanos han confirmado estos hallazgos8-10. Estas aproximaciones hacia la base genética de los trastornos del sensor del calcio no se han trasladado al uso sistemático de pruebas de cribado de mutaciones en pacientes con confirmación bioquímica de la enfermedad. La falta de uso del diagnóstico genético se debe, entre otras razones, a que las pruebas bioquímicas de metabolismo fosfocálcico aportan una distinción clara entre esta afección y el hiperparatiroidismo primario en cualquier momento del período vital del afectado e indican directamente la gravedad de la disfunción del sensor de calcio. Dichas pruebas pueden utilizarse, asimismo, para el cribado de familiares del probando y para el consejo genético, cuando éste se requiera. En conclusión, la fiabilidad del diagnóstico bioquímico ha relegado las pruebas genéticas al terreno de las investigaciones básica y clínica.
En la HHF, en la mayoría de los casos, destaca la ausencia de manifestaciones clínicas. Sólo un pequeño porcentaje de pacientes presenta pancreatitis de repetición o condrocalcionosis3,10. La escasez de manifestaciones clínicas pone el punto de interés del diagnóstico de esta entidad en la distinción del hiperparatiroidismo primario y evitar tratamientos quirúrgicos y/o médicos innecesarios.
El diagnóstico se basa en los datos de laboratorio, que consisten en hipercalcemia moderada e hipermagnesamia leve con una excreción de calcio en orina descendida (inferior a 5mg/kg/día) y PTH discretamente elevada o normal10. Es muy importante el diagnóstico diferencial con el hiperparatiroidismo primario, puesto que este último puede precisar tratamiento quirúrgico en ocasiones. Para distinguir ambas entidades hay que considerar los hallazgos del laboratorio y la historia familiar del paciente11,12. Los siguiente datos orientan a HHF: ausencia de osteopenia, osteítis fibrosa quística, nefrolitiasis y poliuria, aunque algunos pacientes pueden presentar pancreatitis o condrocalcinosis, hipercalcemia en varios miembros de la familia, incluidos niños pequeños, excreción de calcio en orina descendida y ecografía o gammagrafía de paratiroides normales9. El cociente aclaramiento de calcio/aclaramiento de creatinina es muy útil para diferenciar hiperaparatiroidismo primario de HHF. En la HHF suele ser menor de 0,01 y en el hiperparatiroidismo generalmente es mayor de 0,0212. Este cociente se calcula con la siguiente fórmula (calcio urinario×creatinina plasmática) / (calcio plasmático×creatinina urinaria), todos los valores expresados en mmol/l. También es importante descartar otras causas de excreción urinaria de calcio baja, como dieta pobre en calcio y déficit de vitamina D, pero en estas dos situaciones la suplementación con calcio y vitamina D, respectivamente, conduce a hipercalciuria si el paciente tiene hiperparatiroidismo.
El diagnóstico genético no está disponible de forma sistemática y, por tanto, no se efectuó en nuestros pacientes. En la mayoría de los casos revisados en la literatura no hay confirmación genética y el diagnóstico se basa en los hallazgos analíticos compatibles en 2 o más miembros dentro de una misma familia.
La evolución benigna de la HHF hace que no precise tratamiento. La paratiroidectomía no está indicada, salvo en los casos homocigotos o en los pacientes con pancreatitis de repetición. Se recomienda el cribado familiar una vez que se ha diagnosticado un paciente.
En conclusión, la hipercalcemia se presenta con una frecuencia del 5% en los pacientes hospitalizados y en el 1% de la población general. Entre las múltiples causas que pueden producirla la HHF no es frecuente, pero sí es muy importante su diagnóstico para realizar cribado familiar y establecer el diagnóstico diferencial con el hiperparatiroidismo primario.
