


Presentamos el caso de una paciente con clínica de exceso de catecolaminas, con confirmación bioquímica y pruebas de imagen compatibles con paraganglioma. Aunque presentó datos de hiperparatiroidismo e hipergastrinemia que obligaron a descartar una neoplasia endocrina múltiple tipo 1 y 2 (MEN), finalmente se diagnosticó de paragangliomatosis múltiple. Esta entidad se asocia a mutaciones de los genes SDHB-C-D, que en nuestro caso se confirmó la mutación del último. El conocimiento de la genética, mutaciones, tipo de transmisión, vías de tumorogénesis, de estas entidades está en pleno desarrollo, por lo que se presenta una revisión de los últimos datos.
We report the case of a female patient who presented with symptoms of catecholamine excess. Urinary catecholamines were elevated. Radiologic tests were compatible with paraganglioma. High parathyroid hormone and gastrin values were also detected, which excluded multiple endocrine neoplasia type 1 and 2. Finally, the patient was diagnosed with multiple paraganglioma. This entity is associated with mutations in the SDHB-C-D genes. An SDHD mutation was found in our patient. The genetic bases, mutations, genotypephenotype associations, and tumorigenesis of SDH are currently being studied. The present article provides a review of the most recent data.
Los feocromocitomas son tumores de origen neuroectodérmico originados en las células cromafines. Se denominan así por la intensa reacción de tinte producida por la oxidación de los depósitos intracelulares de catecolaminas al exponerse a sales de dicromato. Aquellos que se originan de las células cromafines de la médula suprarrenal se denominan feocromocitomas y los que se originan a partir de los paraganglios se denominan paragangliomas o feocromocitomas extrasuprarrenales.
Los paraganglios son agrupaciones de células de la cresta neural especializadas que han migrado hasta su destino final a través del organismo. Los tumores pueden desarrollarse, por tanto, en los ganglios simpáticos localizados desde el cuello hasta la vejiga, así como en el cuerpo carotídeo, cuerpo vagal, mediastino, aorta, órganos de Zuckerkandl y pelvis (la localización más frecuente). La manifestación clínica más habitual de estos tumores es la hipertensión arterial, mantenida en la mitad de los pacientes y paroxística en un tercio. Puede haber normotensión en una quinta parte de los pacientes1. El diagnóstico se realiza al demostrar una elevación de las catecolaminas o metabolitos en plasma o en orina de 24 horas. Presentamos el caso de una paciente con clínica típica y elevación franca de catecolaminas urinarias.
CASO CLÍNICOMujer de 74 años remitida a consultas de endocrinología por astenia, pérdida de peso y episodios de sudoración y mareo en los últimos meses.
Presentaba como antecedentes personales diabetes mellitus tipo 2 (DM2), hipertensión arterial (HTA), dislipemia y poliposis colónica. Había sido intervenida entre 1984 y 1988 de glomus carotídeo derecho en otro centro hospitalario (no aporta informes) y en 1998 de una tumoración en bifurcación carotídea izquierda. Recibió tratamiento con radioterapia en el año 2003 por presentar recidiva. Seguimiento en cirugía vascular y otorrinolaringología hasta acudir a nuestras consultas en 2006.
Como antecedentes familiares refería que durante su infancia su padre falleció por un accidente y su madre de tuberculosis. No refería enfermedades conocidas en sus hermanos. De sus 5 hijos, una tiene cáncer de cavum y otro cáncer de colon, los demás están sanos.
La paciente refiere desde hace 6 meses episodios de intenso malestar, sudoración, palpitaciones y mareo, sin cefalea, de frecuencia e intensidad crecientes. Refiere cifras de presión arterial (PA) más elevadas de lo habitual, que han precisado modificación -en atención primaria- de su tratamiento farmacológico habitual (se añadió diurético y antagonista del calcio dihidropiridínico al inhibidor de la enzima de conversión de angiotensina [IECA] que tomaba inicialmente), así como empeoramiento de sus controles glucémicos.
En la exploración física destacan cifras de PA de 200/100mmHg y frecuencia cardíaca de 98 lat/min.
En la cabeza y el cuello se palpaba un ligero aumento de la glándula tiroidea, irregular, sin claros nódulos y una masa laterocervical izquierda, de consistencia blanda pero firme, fija. Auscultación cardiopulmonar normal y exploración del abdomen y los miembros inferiores sin hallazgos.
En cuanto a las pruebas complementarias, aportó la siguiente analítica: metanefrina urinaria, 112μg/24h (52-341); normetanefrina, 6.608μg/24h (< 444); noradrenalina plasmática, 37.147pg/ml (< 650); adrenalina plasmática, 65pg/ml (20–60); dopamina plasmática, 2.115pg/ml (< 150); noradrenalina urinaria, 553μg/24h (< 76); adrenalina urinaria, < 4μg/24h (< 1); dopamina urinaria, 104μg/24h (< 390); paratirina intacta (PTHi), 158pg/ml (< 65); calcio (Ca), 10,2mg/dl (< 10,1); fosfato (P), 3,2mg/dl (2,5-5); tirotropina (TSH), 10,65 mUI/ml (0,1-4); tiroxina no unida a proteína (T4l), 0,69ng/dl (0,7-2); anticuerpos antitiroperoxidasa, > 1.000 Ul/l; gastrina, 2.279pg/ml (< 100); somatotropina (GH), 2,7ng/ml (hasta 5); factor de crecimiento insulinoide tipo 1 (IGF-1), 142ng/ml (hasta 250); cortisol libre urinario (CLU), 23μg/24h (75–270); calcitonina, < 9pg/ml (hasta 14); glucohemoglobina (HbA1c), 6,4% (4–6).
Posteriormente se solicitó: insulina basal, 27,5 μU/ml (< 25); glucagón, 88pg/ml (59–177); péptido intestinal vasoactivo (VIP), 15,5 pmol/l (< 30); gastrina, 480,7pg/ml; cromogranina A, 480,7ng/ml (19,4-98,1); calciuria, 122,6mg/24h (< 10,1); 25-OH-vitamina D, 21ng/ml (12–54); 1,25-OH2-vitamina D, 77pg/ml (16–56); TSH, 18,59 mUI/ ml (0,1-4); T4l, 0,59ng/dl (0,7-2).
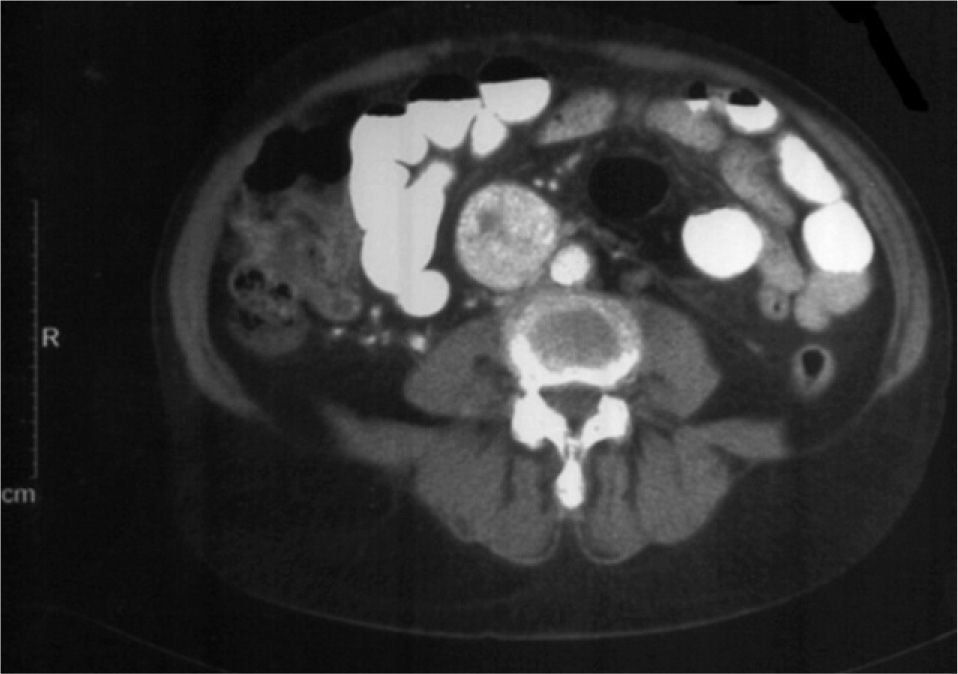
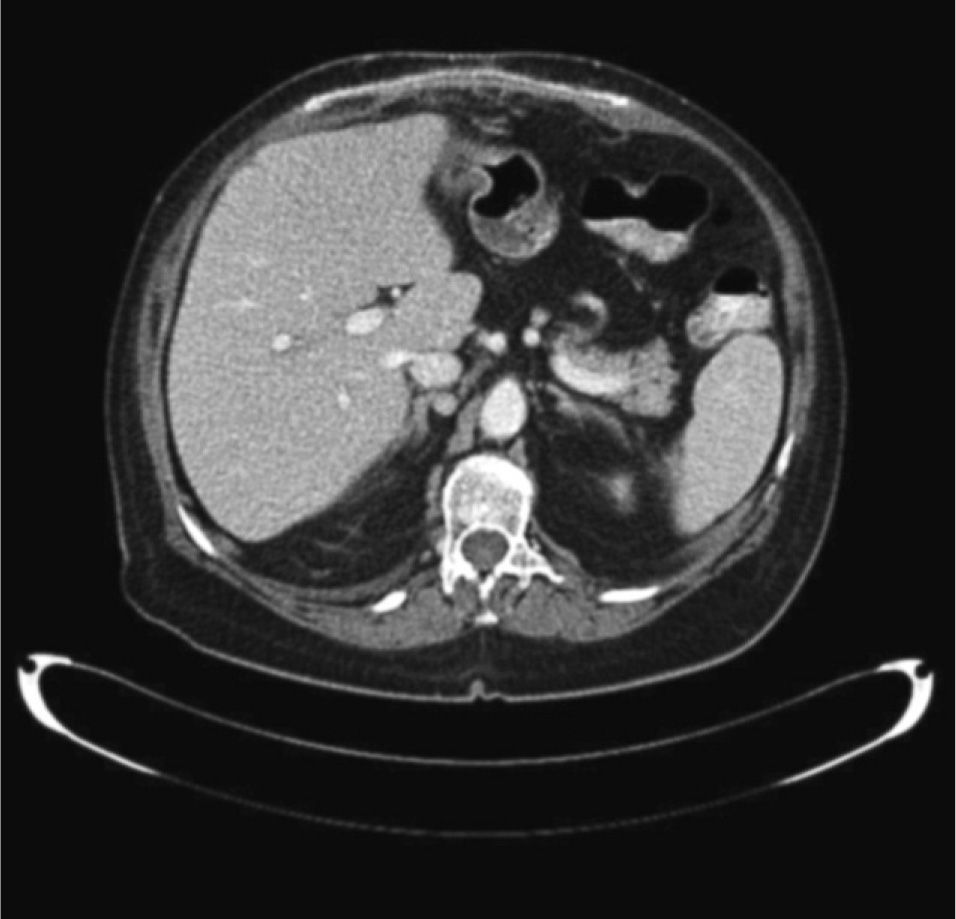
Aportó una tomografía computarizada (TC) de cuello (realizada en 2004) que informaba de lesión hipervascular en bifurcación carotídea izquierda de 4 × 4 × 3cm. Se solicitó una TC abdominal que informó de lesión hipervascular retroperitoneal paraaórtica derecha (fig. 1), compatible con paraganglioma, y otra lesión hipercaptante adyacente a área suprarrenal derecha menor de 1cm (fig. 2).


En la gammagrafía con metayodobencilguanidina (MIBG) se hallaron focos intensamente hipercaptadores en las zonas retroperitoneal y suprarrenal derechas.
La ecografía cervical mostró un probable bocio multinodular (parénquima irregular con patrón micronodular) sin evidencia de alteraciones en el área teórica de las paratiroides, así como una lesión de 3,7cm sólida en bifurcación carotídea izquierda, hipervascularizada.
En la gammagrafía con sestamibi no se observaron captaciones compatibles con adenoma paratiroideo eutópico o ectópico.
La resonancia magnética (RM) de cráneo-hipófisis mostró hallazgos compatibles con microisquemias de larga evolución, sin otras alteraciones.
En el estudio genético se halló una mutación en el exón 2 del gen SDHD.
Ante los hallazgos iniciales de PTH elevada con calcio en el límite alto e hipergastrinemia, se solicitó un estudio completo (bioquímica y de imagen) para descartar asociación de paraganglioma /feocromocitoma con MEN1 o MEN2. Posteriormente se descartó el hiperparatiroidismo primario establecido, los valores sucesivos de Ca y P fueron normales, pese a las cifras elevadas de PTH. Se consideró la hipergastrinemia secundaria a gastritis crónica atrófica observada en gastroscopia solicitada por el servicio de digestivo.
El estudio genético confirmó que la mutación no afectaba al protooncogén RET o al gen de la menina, sino al gen SDHD, lo cual se asocia a paragangliomatosis múltiple en lugar de a las entidades anteriormente referidas.
Tras preparación farmacológica con inhibición alfa y beta y sueroterapia, se realizó la resección quirúrgica de la masa retroperitoneal. Posteriormente, la clínica desapareció, se normalizaron las glucemias y las cifras de PA. La paciente presentó una eventración de la laparotomía. Aunque no se pudo localizar la pequeña lesión parasuprarrenal, las catecolaminas se han mantenido dentro de valores normales, por lo que se mantiene actitud expectante con vigilancia periódica.
DISCUSIÓNAunque la hipertensión arterial es la manifestación más habitual del feocromocitoma/paraganglioma, también es frecuente la aparición de episodios paroxísticos de cefalea, palpitaciones y sudoración2. Más del 90% de los pacientes tienen dos síntomas de la tríada. Otros síntomas menos frecuentes son temblor, angina, náuseas, fenómeno de Raynaud, entre otros. Es frecuente la alteración de la tolerancia a hidratos de carbono o empeoramiento del control glucémico en pacientes diabéticos.
La presión arterial es lábil en estos pacientes por varios factores: liberación episódica de catecolaminas, alteración de reflejos simpáticos y reducción crónica de volumen3. Cuando predomina la secreción de adrenalina, dopa o dopamina, la hipotensión ortostática puede ser el síntoma de manifestación inicial. Puede observarse en ocasiones la aparición de miocardiopatía dilatada o hipertrófica, reversibles tras la resección del tumor.
El diagnóstico se realiza al demostrar una elevación de las catecolaminas o los metabolitos circulantes o en orina de 24 horas. Una determinación plasmática aleatoria puede pasar por alto las concentraciones máximas de catecolaminas. En general, se considera que la determinación de metanefrinas-catecolaminas urinarias es muy útil en la valoración: una elevación 2 o 3 veces por encima de su límite normal es diagnóstica de feocromocitoma. Debe tenerse en cuenta la posibilidad de resultados erróneos si el paciente consume labetalol, antidepresivos tricíclicos, reserpina, clonidina o clofibrato. Igualmente enfermedades como el infarto de miocardio o la enfermedad cerebrovascular aguda pueden elevar las catecolaminas. Para el diagnóstico de localización son de utilidad tanto la TC como la RM; la primera es más sensible y la segunda, más específica. La MIBG tiene similitudes con la noradrenalina, y marcada con 123I o 131I se emplea para la localización gammagráfica, sobre todo, de los feocromocitomas extrasuprarrenales, con una sensibilidad del 90% y una especificidad del 100%4. El OctreoScan es otra técnica de localización, menos específica, dado que estos tumores expresan receptores de somatostatina.
El tratamiento de elección es la resección quirúrgica del tumor. Previamente se realiza inhibición alfa y posteriormente beta (10–14 días) para evitar crisis hipertensivas, así como una adecuada repleción de volumen. En paragangliomas cervicales se ha descrito la realización de embolización selectiva seguida de cirugía como método seguro y efectivo para la resección de tumores, con baja tasa de morbilidad5.
En el caso de nuestra paciente, los datos clínicos eran bastante indicativos de exceso de catecolaminas. En nuestra presencia sufrió crisis de malestar, angustia, con enrojecimiento facial, taquicardia y elevación de PA a 200/100mmHg. Las determinaciones bioquímicas fueron inequívocas para el diagnóstico, y tanto con la TC como con la gammagrafía con MIBG pudo localizarse la procedencia del nuevo tumor productor de catecolaminas. Durante el estudio y el seguimiento de la paciente, la masa cervical no se comportó como hormonalmente activa en ningún momento.
Durante los últimos 10 años se ha reconocido que el feocromocitoma podía ser componente de la enfermedad de Von Hippel-Lindan (VHL), de MEN 2 y neurofibromatosis (NF) 1 en relación con mutaciones germinales en VHL, RET o NF1. En el análisis genético del feocromocitoma esporádico se han detectado mutaciones germinales en el 24% según diversos estudios. En la experiencia de Rotterdam la frecuencia es del 7,5%6.
El síndrome de paraganglioma familiar se ha relacionado con la mutación de los genes SDHD y SDHB, que se transmite de forma autosómica dominante. El locus cromosómico se halla en el cromosoma 11q23, 1p. Se han comunicado deleciones en el punto de empalme, de sentido erróneo y sin sentido. También se han encontrado microinserciones y deleciones. Las mutaciones impiden la producción de proteína funcional. La mayoría de los pacientes tienen historias familiares negativas y no presentan evidencia de enfermedad sindrómica. Los tumores extraadrenales, multifocales y de aparición en la juventud son los que más se asocian a la mutación en la línea germinal. En los paragangliomas esporádicos (sin casos familiares conocidos) se ha detectado mutaciones germinales de SHD en el 28% de los casos, mientras que en casos familiares su presencia es casi invariable7, 8.
Cada vez se conoce más acerca de estos genes relacionados con paraganglioma familiar. Se sabe que codifican proteínas del complejo mitocondrial II de la cadena respiratoria9. Éste es el complejo más pequeño de la cadena y está compuesto por 4 subunidades codificadas por los genes SDHA, SDHB, SDHC y SDHD. Su función es oxidar el succinato-fumarato en el ciclo de Krebs, y está involucrado en la cadena de transporte electrónico respiratoria. SDHA y SDHB codifican flavoproteínas y proteínas ferrosulfuradas, y SDHC y SDHD codifican las dos subunidades hidrófugas transmembrana. Las mutaciones que afectan a la cadena respiratora a menudo se caracterizan por hipotonía, retraso mental, retraso de crecimiento, cardiomiopatía, miopatía, neuropatía, fallo orgánico y alteraciones metabólicas. Estas alteraciones se transmiten vía materna si el defecto genético se ubica en el genoma mitocondrial, mientras que la herencia es mendeliana si se encuentra en el núcleo. Mientras que mutaciones en SDHA causan un fenotipo similar a otros defectos mitocondriales y de Krebs, las mutaciones en SDHB, C y D causan paraganglioma hereditario10.
El paraganglioma familiar se origina de mutaciones germinales heterocigotas inactivantes de SDHB, C y D generando la aparición de tumores de crecimiento lento y muy vascularizados, sobre todo, en el cuerpo carotídeo y el abdomen. En el caso de SDHD, sólo la transmisión vía paterna lleva a la génesis de tumores en la descendencia, pero también se ha observado que el desarrollo de los tumores en portadores se incrementa en altitudes elevadas11.
El mecanismo de acción por el que las mutaciones inducen proliferación tumoral continúa en estudio. Los genes actúan como supresores tumorales en condiciones normales. Tras las mutaciones se ha identificado una sobreexpresión del factor HIF1α (factor inducible por la hipoxia) en relación con un incremento de fumarato-succinato. El HIF1α actuaría sobre factores que favorecen el crecimiento endotelial y aumentan la densidad microvascular. Por tanto parece que el mecanismo básico de la tumorogénesis sería, de forma similar al síndrome de von Hippel-Lindau, la vía seudohipóxica12. También se apunta como determinante la presencia de desequillibrios en la oxidorreducción13.
El estudio clínico de estas entidades no está totalmente desarollado, pero hoy se conocen ya algunos datos importantes. SDHB se asocia a enfermedad extraadrenal y a malignidad. SDHD se asocia a tumores de cabeza y cuello, tumores múltiples, ocasionalmente a feocromocitoma extraadrenal y rara vez a malignidad. Esto es así tanto en el caso índice como en portadores. La edad media al diagnóstico del caso índice es de 34 años. La edad y penetrancia de portadores es mayor en SDHB. SDHD tiene imprinting materno: si la mutación se recibe de la madre no se expresará, pero puede transmitirse, y en el caso de que lo haga un varón, sí puede tener descendencia afectada14.
En general, se recomienda el estudio genético en casos familiares, tumores múltiples, aparición a edades tempranas (50 años)15. Benn et al14 recomiendan el estudio genético de todos los familiares de primer grado tras diagnosticarse el caso índice en mutaciones SDHB y D, porque incluso en sujetos asintomáticos se evidencia, tras estudios bioquímicos o de imagen, la enfermedad latente.
