


La resistencia a hormonas tiroideas es un grupo de síndromes de causa genética caracterizados por la disminución de la sensibilidad tisular a estas hormonas. En la actualidad se distinguen tres formas, en los que la resistencia a la acción hormonal se debe, respectivamente, a mutaciones del gen que codifica el receptor nuclear de T3 TRβ, a alteraciones en el transporte celular de T4 y T3, y a defectos en la conversión de T4 en T3 mediada por desyodasas. En esta revisión se hace una exposición resumida y actualizada de cada una de estas tres formas de resistencia y se discuten los mecanismos patogénicos y aproximaciones clínicas.
Thyroid hormone resistance syndromes are a group of genetic conditions characterized by decreased tissue sensitivity to thyroid hormones. Three syndromes, in which resistance to hormone action is respectively due to mutations in the gene encoding for thyroid hormone receptor TRβ, impaired T4 and T3 transport, and impaired conversion of T4 to T3 mediated by deiodinases. An updated review of each of these forms of resistance is provided, and their pathogenetic mechanisms and clinical approaches are discussed.
La resistencia a la hormona tiroidea fue descrita por primera vez en 1967 por Refetoff et al1,2. Se trata de un síndrome de causa genética caracterizado por la disminución de la sensibilidad tisular a las hormonas tiroideas. La forma clásica se debe en la gran mayoría de los casos a mutaciones en el gen Thyroid Hormone Receptor Beta (THRB) que codifica uno de los dos tipos del receptor nuclear de T3, el Thyroid Receptor β (TRβ). Existe también una forma clínica de resistencia, indistinguible de la forma clásica en la que no existen mutaciones en el TR, y son de causa desconocida. Recientemente también se consideran estados de resistencia a hormona tiroidea a “todos los defectos que interfieren con la actividad biológica de una hormona químicamente intacta secretada en cantidades normales”3. Esta definición comprende también los estados debidos a alteraciones en el transporte celular de T3 y T4, y las alteraciones de la conversión de T4 en T3 mediada por las desyodasas.
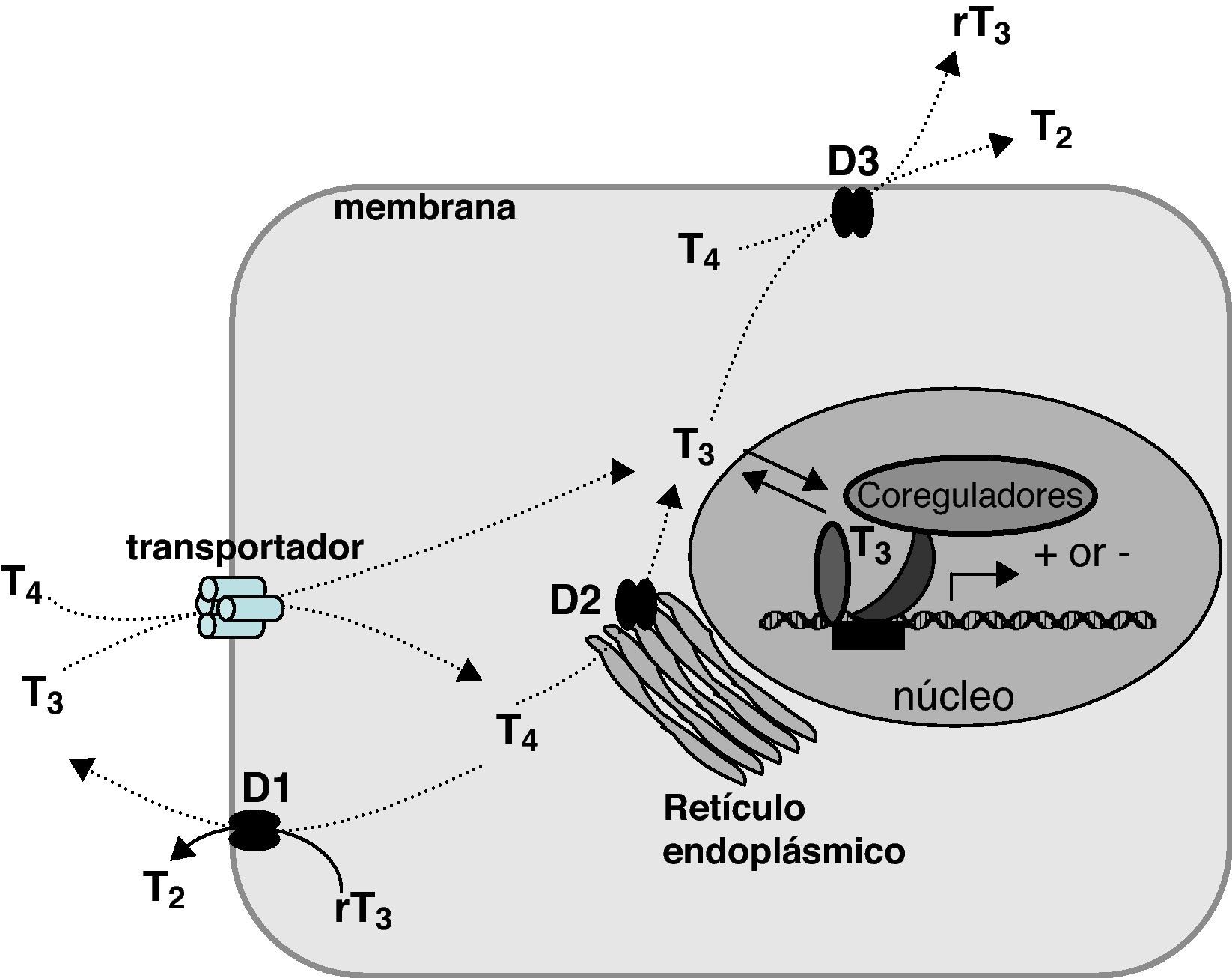
Resistencia a hormonas tiroideas debidas a mutaciones en el receptor TRβMecanismo de acción de hormonas tiroideas (fig. 1)Las hormonas tiroideas regulan aspectos importantes del desarrollo y del metabolismo de casi todos los tejidos de vertebrados. La glándula tiroides segrega dos compuestos hormonales, T4 (tiroxina, 3,5,3’,5’-tetrayodo-L-tironina) y T3 (3,5,3’-triyodo-L-tironina). Esta última también se origina en los tejidos mediante desyodación de T4 catalizada por desyodasas. De hecho, la mayor parte de la T3 en el organismo procede de esta vía. La entrada y salida de las hormonas tiroideas y sus metabolitos a través de la membrana de las células diana requiere la presencia de proteínas de membrana con función de transporte. Aunque existen muchas proteínas transportadoras, la más relevante desde el punto de vista fisiopatológico es Monocarboxylate transporter 8 (MCT8). Esta proteína transporta de manera muy específica T4 y T3, teniendo un papel importante, no sólo en la acción celular de las hormonas tiroideas, sino también en su secreción por el tiroides (fig. 1).
, de monocarboxilatos (MCT8), de aniones orgánicos (OATP) y de Na+/taurocolato (NTCP). La T4 y la T3 son sustratos de las desyodasas, de las cuales D1 y D3 son proteínas de la membrana celular, con el centro activo dirigido hacia el citoplasma, y D2 es una proteína del retículo endoplásmico. La T3 es la hormona activa y se une a receptores nucleares, regulando la transcripción.")
Esquema del metabolismo y acción de las hormonas tiroideas. Se representa la entrada en la célula facilitada por transportadores, de diversas familias de proteínas: transportadores de aminoácidos (LAT-1 y LAT-2), de monocarboxilatos (MCT8), de aniones orgánicos (OATP) y de Na+/taurocolato (NTCP). La T4 y la T3 son sustratos de las desyodasas, de las cuales D1 y D3 son proteínas de la membrana celular, con el centro activo dirigido hacia el citoplasma, y D2 es una proteína del retículo endoplásmico. La T3 es la hormona activa y se une a receptores nucleares, regulando la transcripción.
La hormona tiroidea actúa regulando la tasa de transcripción de genes: la acción fundamental de las hormonas tiroideas se ejerce a nivel del núcleo celular mediante regulación de la transcripción. De esta forma, las hormonas tiroideas controlan la expresión regional y temporal de un gran número de genes que participan en numerosos procesos fisiológicos. La hormona activa a nivel transcripcional es la T3. La T4 es fundamentalmente una prohormona, aunque recientemente se han descrito acciones a nivel de la membrana celular, conocidas genéricamente como “acciones no genómicas”4.
El número de genes regulados por las hormonas tiroideas es muy elevado. En estudios de microarrays comparando tejidos de rata y ratón de animales eutiroideos e hipotiroideos, en un determinado tejido diana como el hígado, se pueden obtener 600-1.000 genes cuya expresión está alterada en el hipotiroidismo. Pero se pueden destacar algunos con especial significado fisiopatológico. En la regulación del eje hipotálamo-hipófisis-tiroides, la TRH hipotalámica y la TSH hipofisaria son reguladas de forma negativa por T3. En ausencia de T3 la transcripción está incrementada, y en presencia de T3 está disminuida. En el corazón, las hormonas tiroideas regulan la contractilidad del miocardio mediante el control de la transcripción de genes como las cadenas pesadas de la miosina y la bomba de Ca2+ del retículo sarcoplásmico (SERCA). En el hígado regula enzimas lipogénicas, como la enzima málica, o del metabolismo del colesterol.
Las acciones genómicas de la T3 se ejercen a través de receptores nucleares: al igual que ocurre con otras hormonas, como los esteroides o la vitamina D, la regulación de la transcripción por T3 se efectúa mediante la interacción con proteínas localizadas en el núcleo conocidas como receptores nucleares. En mamíferos existen dos tipos de receptor de T3, codificados por distintos genes: en el ser humano, el gen Thyroid Hormone Receptor Alpha (THRAα) en el cromosoma 17, codifica el receptor TRα1 y otras proteínas de estructura relacionada, como TRα2, que no unen T3; el gen THRB, en el cromosoma 3, codifica las proteínas receptoras TRβ1 y TRβ2 (abreviadas como TRβ). Estas dos proteínas son idénticas excepto en el extremo amino terminal4.
Los receptores nucleares son factores de transcripción regulables por el ligando hormonal: los receptores tienen una doble función. Por un lado, son capaces de unir la hormona con una alta afinidad, por lo que son sensibles a las bajísimas concentraciones de T3 libre en las células. La unión de la hormona, o ligando, al receptor es reversible y tiene lugar en una parte de la molécula del receptor denominada dominio de unión a ligando o Ligand Binding Domain (LBD). La estructura tridimensional del LBD consiste en una especie de hueco o bolsillo donde se aloja la hormona. Además de la función de reconocimiento del ligando, el receptor tiene una función ejecutiva, lo que lo diferencia de otras proteínas capaces de unir hormona, y se manifiesta mediante su capacidad de interaccionar con el DNA y con otras proteínas nucleares. La unión al DNA ocurre mediante la interacción de una región específica del receptor, conocida como “dominio de unión al DNA” o DNA Binding Domain (DBD), con unas secuencias muy específicas, denominadas “elementos de respuesta”, que en el caso de la T3 se abrevia como Thyroid Response Element (TRE). Otras regiones del receptor, situadas de forma más difusa por su superficie, interaccionan con proteínas nucleares que poseen actividades enzimáticas capaces de reprimir o activar la transcripción. Estas proteínas se denominan “co-reguladoras”, pudiendo ser “co-represoras” o “co-activadoras”, respectivamente5.
Los receptores regulan la transcripción mediante la interacción con otras proteínas nucleares: de forma muy simplificada, en ausencia de T3 el receptor se encuentra unido al DNA mediante el DBD y a proteínas co-represoras, y el gen está silenciado. La unión de T3 al LBD no modifica la unión del receptor al DNA, pero introduce un cambio en la estructura terciaria del receptor, por el cual una de las hélices (la número 12) de la proteína se pliega sobre el hueco donde se aloja la hormona. Este cambio da lugar a la pérdida de la unión con los co-represores, que se disocian del complejo, y a la exposición de una superficie del receptor sobre la que se unen las proteínas co-activadoras. Este esquema se cumple en el caso de genes que son estimulados por la T3, y se habla de “regulación positiva”. Existen también muchos genes cuya expresión es mayor en ausencia que en presencia de T3, por ejemplo el gen que codifica TSH. En estos casos se habla de “regulación negativa”. Aunque en algunos casos el mecanismo funciona de forma inversa a la regulación positiva, es decir, los co-represores actúan como co-activadores y a la inversa, no se conocen bien los mecanismos moleculares implicados. Recientemente se ha demostrado que la regulación positiva de TSH por TRH depende de modificaciones de la histona H4, mientras que la regulación negativa por T3 depende de la histona H3. Un TRβ con una mutación de un paciente con RTH bloqueaba la acción de la T3 sobre la histona H3 impidiendo la regulación negativa por la hormona6.
Los receptores de T3 no se distribuyen de forma homogénea en los distintos tejidos: un aspecto muy importante de la acción de los receptores de T3 es que la actividad transcripcional a nivel molecular y celular de los tres receptores: TRα1, TRβ1 y TRβ2, es muy parecida. Aunque TRβ2 posee algunas propiedades específicas, en general sólo hay diferencias sutiles entre los receptores en cuanto a la afinidad por T3 y a la capacidad de transcribir un determinado gen. Sin embargo, existen grandes diferencias fisiológicas en cuanto al papel de cada una de estas isoformas en el organismo. Estas diferencias son consecuencia de su distribución tisular. Aunque los tres tipos de receptor se expresan en la mayoría de los tejidos, lo hacen en distintas proporciones. En el hígado, más del 80% es TRβ1, mientras que en el cerebro, los huesos, los músculos esqueléticos y el corazón es TRα1 y en la hipófisis es TRβ27. Estos receptores median la acción de T3 en los tejidos mencionados y explican en gran medida muchas de las características clínicas de los pacientes con RTH.
Mecanismos patogénicos en la RTHAlteraciones en los receptores TRβ1 y TRβ2 causan resistencia a las hormonas tiroideas: la mayoría de los casos de resistencia a hormonas tiroideas se deben a mutaciones en el gen THRB (fig. 2) que, como hemos dicho, codifica TRβ1 y TRβ28. Las mutaciones de TRα1 en el ser humano están aún por descubrir. La deleción completa del gen THRB, con ausencia de TRβ sólo se ha encontrado en la familia original descrita por Refetoff et al9. Existen dos clases de mutaciones: aquellas que disminuyen la afinidad por T3 y otras que, manteniendo la afinidad por la hormona a un nivel normal, hacen que el receptor interaccione de forma anómala con las proteínas nucleares co-reguladoras. Desde el punto de vista fisiopatológico está alterada la respuesta de los órganos que expresan TRβ1 y TRβ2. Como consecuencia de la mutación en TRβ2 la sensibilidad de la hipófisis a T3 disminuye, por lo que se necesitan concentraciones más elevadas de T3 circulante para inhibir la TSH. Lo mismo ocurre a nivel hipotalámico en cuanto a la regulación de TRH. El eje TRH-TSH-tiroides se mantiene regulado a un set point mayor, coexistiendo concentraciones elevadas de T4 y de T3 con TSH no suprimida. En órganos como el hígado o los riñones, donde la acción de T3 se ejerce principalmente a través de TRβ1, las concentraciones hormonales en el paciente con resistencia son adecuadas a la mutación presente. Por el contrario, en órganos donde la acción de T3 se ejerce a través de TRα1, como el corazón y los huesos, el incremento de T3 ocasiona un estado de hipertiroidismo local, con taquicardia, arritmias y trastornos de la mineralización, respectivamente.
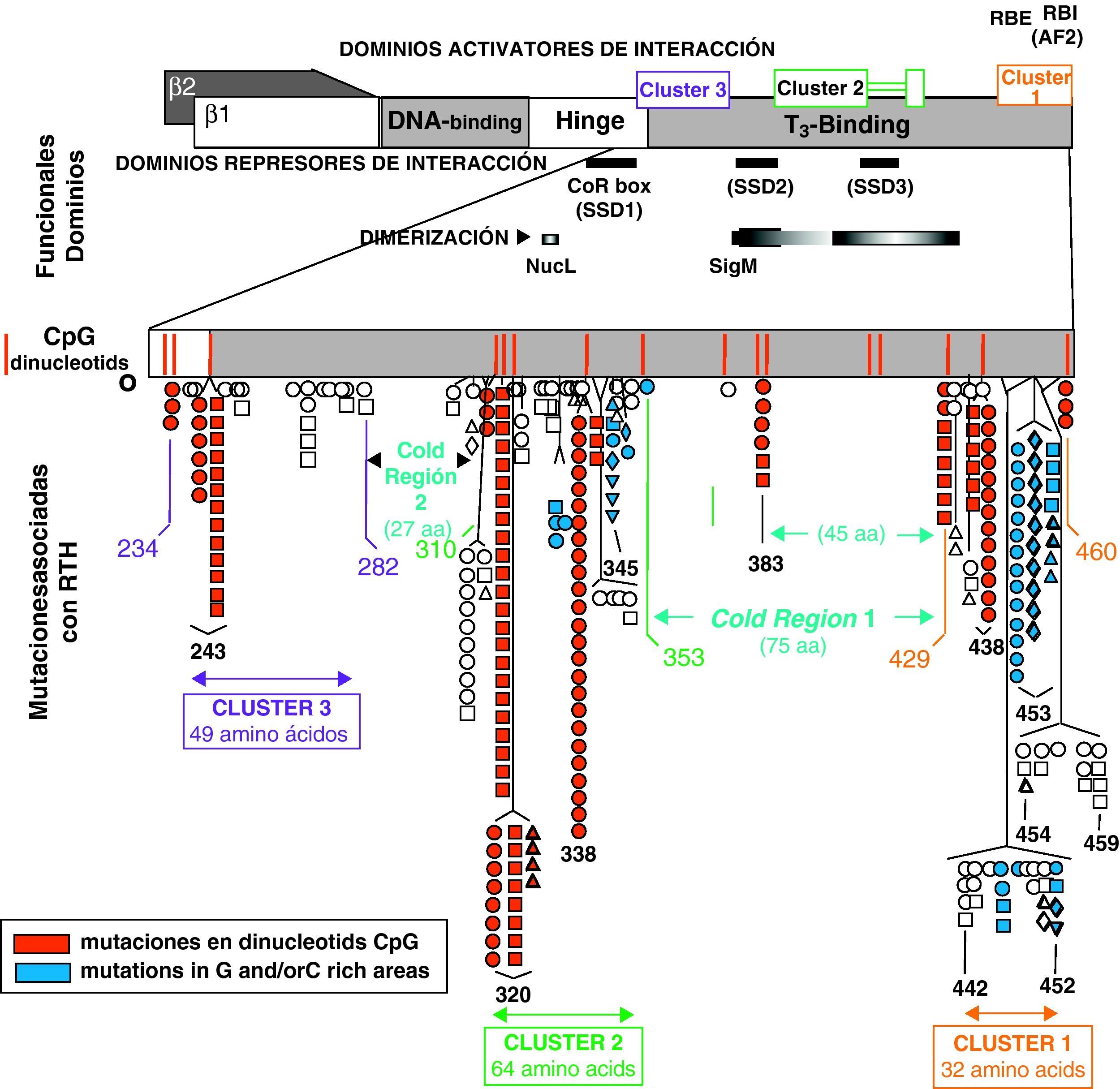
 y de dimerización. Las mutaciones se agrupan en 3 zonas (Cluster 1, 2 y 3), en regiones ricas en dinucleótidos CpG. (Reproducido con permiso de: Refetoff D. The syndromes of resistance to thyroid hormones. En: DeGroot LJ, editor. Thyroid Disease Manager. Capítulo 16. Figura 3. Disponible en: http://www.thyroid manager.org).")
Estructura de los receptores TRβ1 y TRβ2, con los dominios funcionales de unión a DNA y a T3. También se indican las zonas de la molécula que interaccionan con los co-represores (SSD1, SSD2 y SSD3) y de dimerización. Las mutaciones se agrupan en 3 zonas (Cluster 1, 2 y 3), en regiones ricas en dinucleótidos CpG. (Reproducido con permiso de: Refetoff D. The syndromes of resistance to thyroid hormones. En: DeGroot LJ, editor. Thyroid Disease Manager. Capítulo 16. Figura 3. Disponible en: http://www.thyroid manager.org).
Una misma mutación puede dar lugar a distintas formas clínicas: en algunos casos, aunque TRβ1 y TRβ2 presenten la misma mutación, la actividad de cada receptor no se afecta en la misma cuantía, existiendo un mayor grado de resistencia a nivel hipofisario. La consecuencia es que la resistencia se presenta sólo a nivel hipofisario y el resto del organismo padece exceso hormonal. La descripción de los primeros casos con estas características, es decir, hipertiroidismo con TSH elevada no debido a tumor secretor de TSH10 propició la distinción entre “resistencia generalizada” y “resistencia hipofisaria o central” como entidades distintas. Actualmente se considera que no se trata de síndromes distintos, sino de formas distintas de expresión clínica con un origen molecular común, siendo manifestaciones extremas de la misma enfermedad11. La misma mutación puede dar lugar a cada una de estas dos formas clínicas en distintos pacientes. Precisamente, uno de los problemas fisiopatológicos más complejos que plantea la RTH es por qué la misma mutación puede dar lugar a distintas formas clínicas entre familias o incluso en pacientes pertenecientes a la misma familia12,13.
La causa de la mayor resistencia de la hipófisis en determinados casos no se conoce con exactitud. Es posible que sea una situación extrema de las diferencias de sensibilidad a la T3 que pueden presentar los distintos órganos en el mismo paciente14. En un intento de aclarar el mecanismo, se analizó el efecto de expresar en ratones una mutación en TRβ que produce resistencia central en humanos15,16. Los ratones presentaron TSH elevada y signos de hipertiroidismo, es decir, una resistencia aparentemente limitada a la hipófisis. Esto haría pensar que la función anormal del receptor mutado sólo se manifiesta en la hipófisis, manteniéndose una respuesta normal a T3 en el resto de los tejidos. Sin embargo, se observó que el receptor mutado había perdido la capacidad de regulación negativa de la transcripción en todos los tejidos, manteniéndose intacta la capacidad de regulación positiva, también en todos los tejidos. Es decir, el efecto de la mutación no se limitaba a la hipófisis, como cabía deducir del fenotipo de los ratones. El problema es que en hipófisis la pérdida de la regulación negativa en la célula tirotropa tiene como consecuencia un incremento de la secreción de TSH y, por consiguiente, estimulación tiroidea. La pérdida de regulación negativa por el receptor mutado en otros tejidos u órganos como el hígado, los riñones, los músculos y los huesos tiene menos consecuencias, siendo más relevante fisiológicamente la regulación positiva. Debido a ello, los tejidos citados continúan siendo sensibles al exceso de hormonas tiroideas, apareciendo síntomas de hipertiroidismo.
Las mutaciones se agrupan en zonas calientes de la molécula del receptor: se han descrito 124 mutaciones en alrededor de 350 familias; es decir, la misma mutación está presente en varias familias. Por ejemplo, la mutación R338W está presente en 29 familias no relacionadas entre sí. En el 95% de los casos las mutaciones consisten en sustituciones de un solo nucleótido, que dan lugar a cambios en un aminoácido, o en unos pocos casos a un codón de terminación. Otras alteraciones genéticas de TRβ consisten en deleciones o inserciones con cambios en la pauta de lectura. La mayoría de las mutaciones se localizan en 3 regiones ricas en dinucleótidos CpG (llamadas islas de CG o zonas calientes) presentes en la región carboxilo terminal de TRβ, y afectan al dominio de unión a T3 y de dimerización con otras proteínas nucleares (fig. 2).
El receptor mutado posee actividad dominante negativa: en principio, es fácil concebir que una mutación que afecte a la zona de unión a la hormona produzca un estado de resistencia, de forma que la disminución de afinidad del receptor haga necesaria una mayor cantidad de hormona para producir el efecto biológico. Sin embargo, la realidad es más compleja, pues en muchos casos no existe correlación entre la afinidad del receptor por la T3 y la gravedad de los síntomas17.
En la única familia descrita con deleción completa de TRβ, de herencia recesiva, sólo los pacientes homocigotos presentaban manifestaciones clínicas; es decir, que en los individuos heterocigotos el producto del otro alelo TRβ, junto al TRα normal, era capaz de suplir la falta de uno de los alelos TRβ. Esto no ocurre en el resto de los pacientes con mutaciones puntuales del receptor, de herencia dominante, en los cuales la presencia de un solo alelo mutado es capaz de producir la sintomatología de la RTH. Es decir, el receptor mutado es capaz de inhibir la actividad de los receptores normales, lo que se conoce como acción dominante negativa.
La ausencia de receptor es menos perjudicial que la ausencia de hormona: la acción dominante negativa del receptor mutado se debe a la propiedad de los receptores de T3 de poseer actividad en ausencia de ligando. En los genes regulados de forma positiva por la hormona tiroidea, en ausencia de hormona el receptor es un potente represor de la transcripción, mientras que en los genes regulados negativamente el receptor, en ausencia de hormona, tiene actividad de inducción de la transcripción. Así pues, la falta de hormona no es equivalente a la falta de receptor, sino mucho más perniciosa como hemos demostrado en animales experimentales. En ratones con deleción de TRα1 el hipotiroidismo experimental produce menos alteraciones que en ratones normales18. También en ratones, la deleción de TRβ produce un estado de RTH19. Sin embargo, la mutación en TRβ1 produce, además, graves alteraciones estructurales y funcionales del cerebelo, debido a la actividad dominante negativa del receptor mutado.
Para la actividad dominante negativa se requiere la unión del receptor mutado al DNA: uno de los receptores mutados más potentes, el aislado de la familia S20,21 (deleción de treonina 332) es incluso capaz de inhibir la respuesta transcripcional al ácido retinoico y a la vitamina D322. La acción dominante negativa es posible gracias a que la unión al DNA está intacta. De hecho, la introducción de mutaciones en esta zona no induce actividad dominante negativa o la suprime en un receptor previamente mutado23. Para que se manifieste la actividad dominante negativa es necesario, además, que los dominios de dimerización estén intactos. El receptor mutado sería capaz, pues, de formar heterodímeros sobre el DNA, produciendo represión aun en presencia de hormona e interfiriendo con la activación de los receptores normales mediada por T3. La falta de unión de T3 haría que el receptor estuviese de forma permanente formando un complejo represor.
Algunas mutaciones alteran la interacción del receptor con proteínas nucleares co-activadoras o co-represoras: en la mayoría de los casos de RTH, la actividad dominante negativa es debida a que la falta de unión de T3 al receptor impide la liberación de los co-represores y unión de los co-activadores. Sin embargo, existen casos en que la unión de T3 al receptor mutado no se altera. En estos casos, la resistencia se debe a interacción defectuosa del receptor mutado con los co-represores o los co-activadores. Se han descrito mutaciones que confieren al receptor distintas propiedades en este sentido: 1) existen mutaciones que confieren un incremento de la afinidad de receptor por los co-represores24; 2) otras mutaciones se caracterizan por producir una disociación lenta del co-represor; 3) el tercer grupo de mutaciones origina una unión defectuosa de los co-activadores25. 4) finalmente, algunas mutaciones modifican de forma selectiva la interacción con algunos co-activadores y no otros, introduciendo heterogeneidad en las presentaciones clínicas en función de la distribución tisular de los distintos co-activadores. El caso descrito por Wu et al26 es de gran interés porque ilustra cómo una mutación puede cambiar la selectividad de interacción del receptor con las moléculas co-reguladoras. Las características clínicas del síndrome resultante dependerían en gran medida de la expresión diferencial de estas moléculas en distintos tejidos. En el caso citado, el síndrome era de manifestación casi exclusivamente hipofisaria. Es fácil deducir la gran variedad posible de formas de presentación clínica.
Síndrome de resistencia no-TR: en aproximadamente un 15% de los casos de resistencia no se ha podido demostrar la presencia de mutaciones en el receptor. Se conocen como resistencias no-TR y se describieron por vez primera por Weiss et al27. Se sospecha que en estos casos podría haber mutaciones en alguno de los muchos componentes que interaccionan con el receptor directa o indirectamente y que forman parte del complejo agregado de proteínas co-reguladoras que finalmente interacciona con la maquinaria transcripcional. La atención inmediata se ha dirigido a la búsqueda de mutaciones en las proteínas co-represoras o co-activadoras. En ratones, la deleción del co-activador SRC-1 ocasiona un estado de resistencia a hormonas tiroideas28. Sin embargo, los esfuerzos en demostrar alteraciones en co-represores o co-activadores en humanos han resultado baldíos hasta el momento29. Hay que tener en cuenta la posibilidad de mosaicismo, es decir, cuando el receptor mutado no está presente en todos los tejidos30 y la detección de la mutación depende de si está presente o no en las células sanguíneas. En muchos casos se ha descartado esta posibilidad mediante análisis de ligamiento29.
Características clínicasLa RTH es una enfermedad genética autosómica de baja incidencia: la RTH es de carácter familiar, aunque alrededor del 28% de las mutaciones ocurre de novo. Se han descrito algo más de 600 casos, en unas 350 familias, con una incidencia probable de 1/50.000. En un estudio realizado en Japón en 83.232 recién nacidos, utilizando datos del cribado neonatal, se encontraron 11 niños con T4 elevada. De éstos, un caso de hipertiroxinemia disabulminémica familiar, 2 de RTH y 8 de enfermedad de Graves neonatal31. El caso original descrito por Refetoff et al1, debido a la deleción de TRβ, era de herencia recesiva, manifestándose el síndrome sólo en individuos homocigóticos. Éste ha sido el único caso descrito de RTH por deleción de TRβ. Todos los casos descritos posteriormente son de carácter dominante. Un caso en que la mutación estaba presente en los dos alelos ocasionó una forma extremadamente grave de resistencia21. Se ha descrito recientemente un caso de mosaicismo, con la mutación presente sólo en algunos tejidos30.
En la RTH coexisten signos de deficiencia y exceso hormonal: una descripción exhaustiva de las características clínicas fue publicada hace algunos años en una revisión muy completa32. También se pueden consultar descripciones más recientes3,8,33. El hallazgo esencial en estos pacientes es la presencia de concentraciones altas de hormonas tiroideas circulantes (T4 y T3 libres) en presencia de TSH normal o ligeramente elevada. Como hemos dicho anteriormente, esta situación es debida a una disminución de la sensibilidad de la célula tirotropa hipofisaria a la acción de la T3 en la inhibición de la producción de TSH, y de las neuronas del núcleo paraventricular hipotalámico en la producción de TRH. El aumento de la secreción de TSH produce estimulación de la glándula tiroides, siendo el bocio el signo más frecuente, presente en el 95% de los pacientes. El aumento de la secreción de hormonas tiroideas produce un estado de hipertiroidismo en los pacientes con resistencia limitada a la hipófisis. En los pacientes con RTH generalizada los pacientes pueden mantenerse en una situación de eutiroidismo. Sin embargo, es frecuente encontrar manifestaciones clínicas compatibles con defecto o exceso hormonal que afectan al corazón, el sistema nervioso central y al desarrollo. En un elevado número de pacientes se encuentra taquicardia, debida a la acción del exceso de hormona a través de TRα1, presente en el corazón. Algunos pacientes, aún con frecuencia cardíaca normal, presentan síntomas similares a las del hipotiroidismo34. En individuos adultos la densidad mineral ósea es normal en la columna vertebral, pero está significativamente disminuida cuando se mide en el cuello del fémur, por lo que la osteoporosis puede ser un problema importante en la RTH. Estudios mediante biopsia sugieren una menor tasa de formación ósea.
Signos de alteración del desarrollo consisten en estatura baja, retraso de la dentición y de la edad ósea, y sordera. En cuanto al sistema nervioso central, el síndrome de resistencia en niños se asocia con frecuencia a trastornos del aprendizaje, con retraso escolar, alteraciones del lenguaje e incluso deficiencia mental con cociente intelectual bajo en el 5-10% de los casos. Un 70% de los niños con RTH presenta un cuadro de hiperactividad y déficit de atención (Attention Deficit Hyperactivity Disorder o ADHD). Este síndrome está también presente en un 50% de los adultos con RTH. Esta frecuencia es mayor que en la población general: 20 y 7%, respectivamente, por lo que se ha llegado a sugerir que el síndrome de ADHD estaría ligado genéticamente a mutaciones del receptor de T3. Sin embargo, no existe un aumento de la incidencia de RTH en sujetos de ADHD en relación con la que presenta la población en general. La asociación de RTH a un cociente intelectual bajo podría aumentar la probabilidad de síntomas de ADHD35.
DiagnósticoEl síndrome de RTH debe sospecharse en individuos que presentan concentraciones altas de hormonas tiroideas en suero, junto a una TSH normal o elevada, especialmente en presencia de bocio. En suero, tanto la T4 como la T3, total y libre, están elevadas. El mínimo requerimiento para el diagnóstico es una elevación real de la T4 libre en presencia de TSH no suprimida. El diagnóstico diferencial debería plantearse con el hipertiroidismo debido a enfermedad de Graves o a bocio autónomo uninodular o multinodular, la presencia de un adenoma hipofisario secretor de TSH y las anomalías de las proteínas transportadoras de hormonas tiroideas.
Dada la presencia de bocio con algunos signos de hipertiroidismo, como taquicardia y, a veces, hiperactividad, es relativamente frecuente el diagnóstico de enfermedad de Graves, e incluso que se apliquen tratamientos agresivos, como la tiroidectomía parcial, obviamente sin ningún resultado dado que, además, el bocio recurre en poco tiempo. La detección de concentraciones basales no suprimidas de TSH en la RTH y la presencia de anticuerpos circulantes en la enfermedad de Graves pueden ayudar a establecer el diagnóstico. No obstante la RTH y la enfermedad de Graves pueden estar presentes en el mismo paciente36. Estos pacientes requieren dosis elevadas de hormona tiroidea para mantener el eutiroidismo tras la ablación tiroidea. En los adenomas secretores de TSH existe un incremento de la subunidad alfa de la misma y el diagnóstico de adenoma se establece tras la exploración radiológica.
Las alteraciones de las proteínas transportadoras pueden ser confundidas erróneamente con RTH: las alteraciones de las proteínas transportadoras pueden dar lugar a aumento de la T4 sérica total en presencia de una TSH normal. Especialmente hay que tener en cuenta la hipertiroxinemia disalbuminémica familiar37, debida a la presencia de una albúmina circulante con elevada afinidad por la T4. En condiciones normales la albúmina transporta hasta el 10% de la T4 sérica, mientras que en los casos de disalbuminemia puede llegar a transportar el 30%, elevándose la T4 total pero manteniéndose normal la T4 libre. También se han descrito alteraciones de la transtiretina, que se han diagnosticado erróneamente como hipertiroidismo o como RTH38. Las alteraciones de las proteínas transportadoras no suelen cursar con síntomas y, generalmente, existe un aumento selectivo de la T4 total, manteniéndose la T4 libre, así como la T3 total y libre, dentro de los límites normales. Pueden existir casos ambiguos en los que la presencia de bocio debida a otras causas, y de un perfil hormonal no demasiado claro, puede dar lugar a problemas de diagnóstico. Especialmente si se determina la fracción de T4 libre por los métodos que usan un análogo de la T4, ya que, debido a la presencia de proteínas transportadoras anómalas, los resultados son anormalmente altos. En estos casos es necesario recurrir a determinaciones especiales, como la medida de hormona libre mediante diálisis de equilibrio y la electroforesis del suero tras incubación con T4 marcada. Estas determinaciones, no obstante, suelen estar fuera del alcance del laboratorio clínico, por lo que se puede descartar la RTH observando cuidadosamente la concentración de T3 total, que suele ser normal, o bien realizando un test de TRH tras el tratamiento con T3, como se describe en el párrafo siguiente.
La respuesta a la administración de T3 está reducida en los pacientes con RTH: el diagnóstico clínico de RTH se establece evaluando la respuesta a la administración de T3, mediante el protocolo descrito por Refetoff et al32. Éste consiste en la administración de dosis diarias de T3 de forma gradual en tres períodos de una semana: 50, 100 y 200 μg/día en adultos, divididos en dos tomas. Estas dosis se ajustan para los niños en función de la edad y el peso: una dosis de 100 μg para un adulto equivale a 25 μg en niños de 8-15 kg (1-3 años), 50 μg en niños de 16-25 kg (4-9 años) y 75 μg en niños de 26-45 kg (10-14 años). Antes del tratamiento y al final de cada semana se realiza un test de TRH. En individuos normales, ya la dosis de 50 μg de T3 consigue disminuir la respuesta hasta casi suprimirla. En individuos con RTH se necesitan dosis muy superiores, pudiéndose obtener incrementos de TSH aun con la dosis más elevada8. Si, además de TSH, se determinan otros parámetros de acción de T3, como colesterol, ferritina, creatina fosfoquinasa, excreción de hidroxiprolina o la concentración de globulina transportadora de hormonas sexuales (SHBG), se puede apreciar que mientras que en el individuo normal ya existen respuestas con la dosis inferior de T3, en la RTH se necesitan las dosis altas y las respuestas son muy inferiores a las del individuo normal. El diagnóstico final puede confirmarse mediante la secuenciación de TRβ. Dada la facilidad con la que es posible secuenciar TRβ, resulta práctico recurrir a esta prueba antes de aplicar el protocolo de Refetoff et al, que puede ser innecesario si se detecta una mutación, especialmente si la mutación ha sido previamente descrita como patogénica. En el laboratorio especializado es sencillo comprobar si una determinada mutación es patogénica analizando su actividad en líneas celulares. Es preciso tener presente que el 15% de los pacientes no presenta mutaciones en el receptor, por lo que la identificación de mutaciones confirma la resistencia, pero su ausencia no la descarta.
Aunque la mayoría de los pacientes no requiere tratamiento, hay que tener en cuenta situaciones especiales: los pacientes con RTH que presentan un estado de eutiroidismo compensado no necesitan tratamiento y maniobras tendentes a reducir las hormonas circulantes deben ser evitadas3. Aunque en la mayoría de los casos el bocio es moderado, a veces puede ser de un tamaño que requiere tratamiento. No se debe recurrir al tratamiento quirúrgico. Se han conseguido buenos resultados administrando dosis elevadas de T3 cada dos días39. En los casos con signos de hipotiroidismo tisular, por ejemplo retraso del desarrollo en niños o hipercolesterolemia en adultos, es necesario el tratamiento con T4, incluso a dosis altas, usando como índice de actividad biológica la normalización de las concentraciones de TSH y otros índices de acción periférica40. La taquicardia se trata con bloqueantes beta-adrenérgicos como atenolol. Los casos de resistencia restringida a la hipófisis, con signos de hipertiroidismo periférico, plantean problemas especiales. En estos casos es necesario disminuir los niveles de hormonas tiroideas circulantes sin producir una elevación crónica de TSH, por el riesgo de producir una hiperplasia de las células tirotropas. Los agentes dopaminérgicos son útiles, aunque pierden su efectividad cuando se administran de forma crónica. En ciertos casos, el derivado acético de la T3, el TRIAC (ácido 3,5,3’-triyodo-tiroacético) se ha usado con cierta efectividad26,40,41, pues tiene mayor poder de inhibición de la secreción de TSH que de estimulación del metabolismo periférico.
El manejo de pacientes con RTH durante el embarazo plantea problemas especiales: en función de la presencia o no de la mutación en el feto se ha observado menor viabilidad de fetos normales o menor peso al nacer, debido probablemente al exceso de hormona materna42. Los fetos con la misma mutación que la madre estarían protegidos del exceso hormonal, pero no así los fetos normales. El tratamiento de las madres con RTH asintomática no requeriría tratamiento43. Sin embargo, se ha sugerido determinar el genotipo del feto. En caso de que la mutación esté presente también en el feto, no es necesario el tratamiento. Si el feto es normal, se ha recomendado mantener la T4 libre materna por debajo del 20% del límite superior de la normalidad mediante tratamiento muy controlado con agentes antitiroideos3. Naturalmente, hay que tener un cuidado especial de no producir hipotiroidismo fetal.
Alteraciones del transporte celular de hormonas tiroideasLas mutaciones en el transportador de hormonas tiroideas MCT8 producen graves alteraciones neurológicas: En 2004 dos grupos describieron de forma independiente en varias familias un síndrome neurológico y retraso mental en niños ligado al cromosoma X44,45. El cuadro neurológico consistía en retraso del desarrollo psicomotor, retraso mental profundo, falta de comunicación verbal, hipotonía axial y control deficiente de la postura de la cabeza combinada con espasticidad de los miembros que llegaba a producir tetraplejía. Los pacientes presentaban alteraciones de las hormonas tiroideas circulantes consistentes en disminución de T4 y rT3 y aumento de T3. La TSH era normal o ligeramente elevada. El cuadro hormonal sugería un trastorno en la distribución y metabolismo de las hormonas tiroideas, y se encontraron mutaciones en una proteína, denominada MCT8, codificada por el gen SLC16A2 o Solute carrier family 16 member 2, en el locus Xq21) cuya función conocida es la de transportar T4 y T3 a través de la membrana celular46.
Un año más tarde, en 2005, Schwartz et al47 encontraron mutaciones en MCT8 en pacientes de varias familias aquejadas de un síndrome neurológico y de retraso mental ligado al cromosoma X, ya descrito en 194448, el síndrome de Allan-Herndon-Dudley (código de Orphanet ORPHA59), de etiología desconocida. Hasta la fecha hay descritas unas 40 familias. Se ha descrito también mutaciones de MCT8 en el 4% de pacientes con el síndrome de Pelizaeus-Merzbacher49, una leucodistrofia que, en su forma clásica, se debe a mutaciones de la proteína de mielina PLP1 (proteína proteolipídica).
Características clínicasEl síndrome se caracteriza por dos componentes bien diferenciados, endocrino y neurológico8,50–55. El perfil de las hormonas tiroideas circulantes es muy característico e inusual, con elevación de T3 y disminución de T4 y rT3. La TSH puede estar ligeramente elevada, pero en la mayoría de casos es normal o está en el límite superior de la normalidad y tiene poco valor diagnóstico. Además de las alteraciones endocrinas existe daño neurológico56, retraso mental y del desarrollo ligado al cromosoma X. Por lo tanto, se dan en niños, y las mujeres son portadoras. Existe un único caso descrito de afectación neurológica en una mujer, probablemente debido a inactivación sesgada del cromosoma X57.
Al nacer, los pacientes tienen un aspecto normal. En uno de los casos, la T4 del cribado neonatal estaba ligeramente disminuida, pero la TSH era normal. En la mayoría de los casos el síndrome comienza a manifestarse en los primeros meses de vida, con hipotonía truncal e incapacidad de sostener la cabeza, sentarse o gatear. La hipotonía está presente en el 100% de los casos. Existe retraso global del desarrollo y falta casi total de adquisición del lenguaje. La hipotonía progresa a espasticidad con el tiempo. El retraso mental es profundo y de hecho el cociente intelectual se mantiene por debajo de 3047. Otros signos son la postura anormal de las manos, facies alargada y constitución asténica y consunción debida a hipertiroidismo periférico. En niños son frecuentes los movimientos anormales consistentes en extensiones de los miembros de un lado y flexión simultánea contralateral con rotación de la cabeza, que pueden dominar el cuadro clínico55, y episodios de discinesias paroxísticas en lactantes provocados por cualquier estímulo58.
FisiopatologíaPara el estudio de la fisiopatología de este síndrome se han generado ratones modificados genéticamente, en los cuales se ha delecionado uno de los exones del gen, produciéndose una proteína no funcional59,60. Los ratones presentan el cuadro endocrino pero desgraciadamente no presentan alteraciones neurológicas. Esto hace que sólo sean un modelo parcial de la enfermedad.
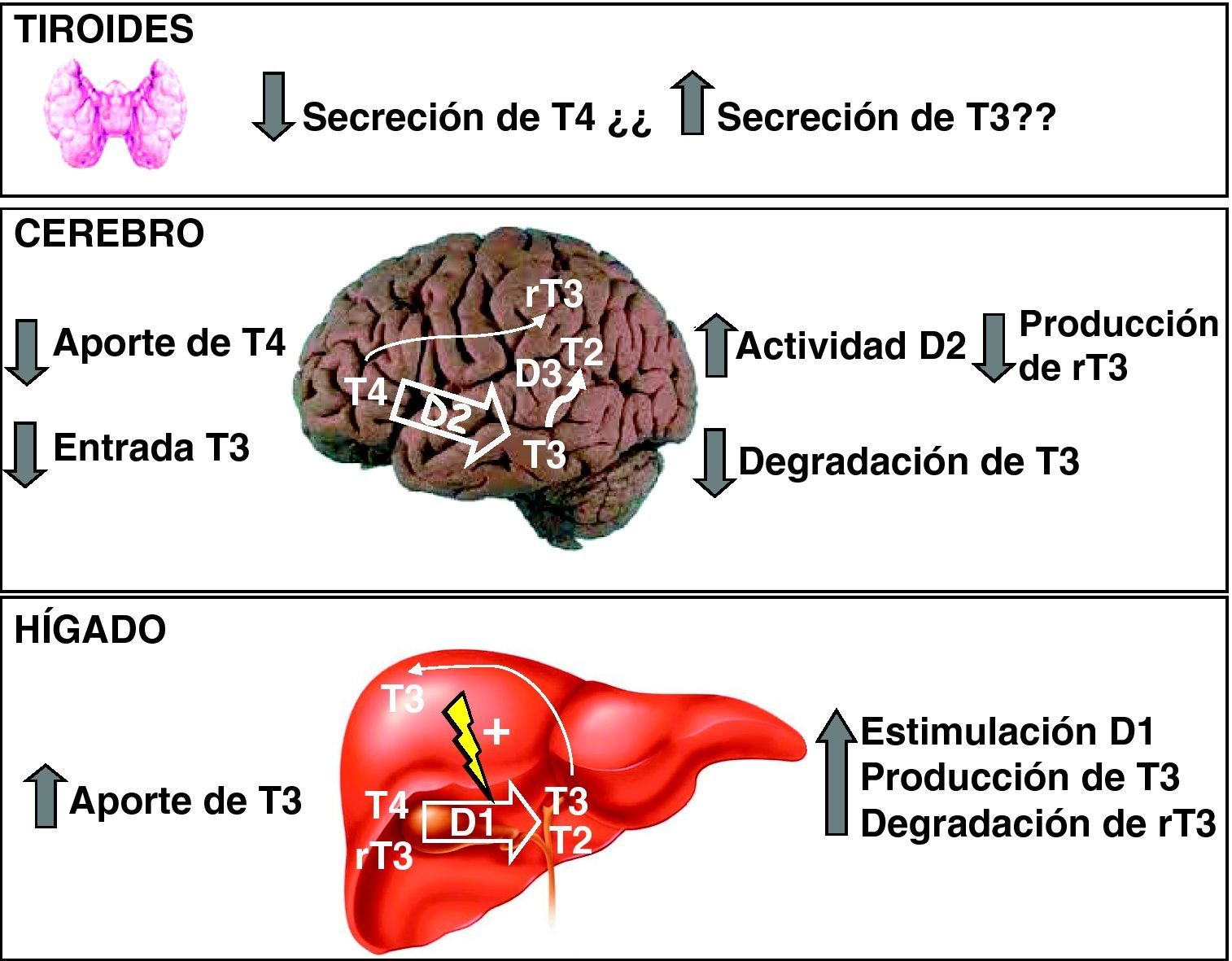
Del estudio de estos ratones se han sacado las siguientes conclusiones: Mct8* es una proteína esencial para el transporte de T3 al cerebro, principalmente por su localización en la barrera hematoencefálica61,62. En ausencia de Mct8 la T3 no llega en cantidad suficiente al cerebro (fig. 3). Aunque Mct8 también transporta T4, ésta también puede atravesar la barrera por medio de otro transportador, el Organic anion transporter polypeptide 14 (Oatp14), producto del gen Solute carrier organic anion transporter family 1c1 o Slco1c1. Mct8 también se expresa en la célula tiroidea y participa en la secreción de T4 y T363,64. En ausencia de Mct8 se acumulan T4 y T3 en tiroides, con disminución de la secreción de T4 y posiblemente, aún en discusión, aumento de la secreción de T3 por mecanismos no bien perfilados. Los ratones knock out de Mct8 también presentan un incremento notable de la actividad desyodasa 1 (D1) en el hígado y los riñones. En general, los ratones presentan hipotiroidismo cerebral, aunque compensado parcialmente, e hipertiroidismo periférico, lo que es la causa de la emaciación que sufren los pacientes.
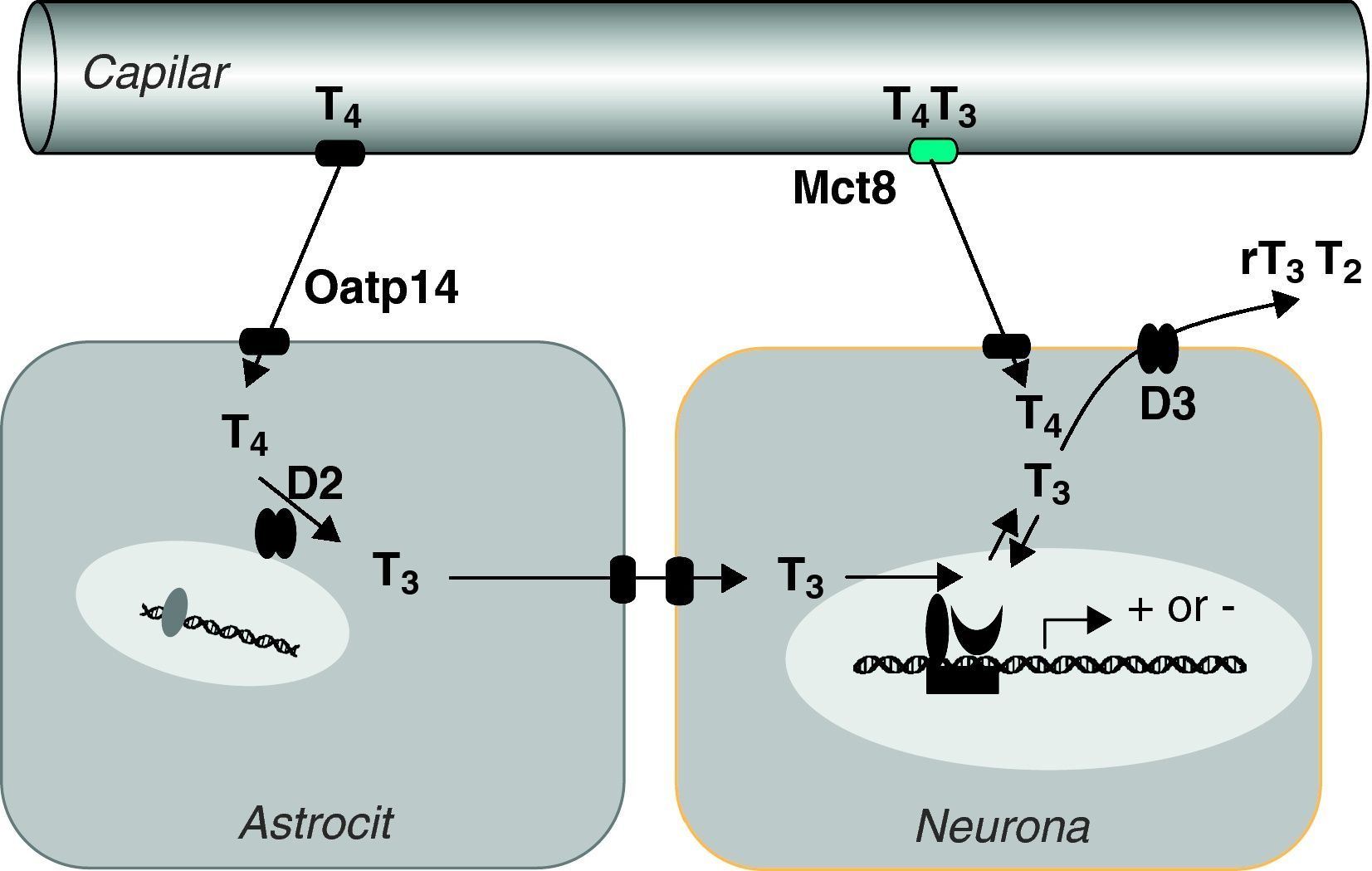
. En la barrera hematoencefálica de roedores el transportador Oatp14 facilita el paso de T4 a los astrocitos, donde se convierte en T3. Mct8 facilita el paso de T4 y T3 probablemente al espacio intersticial, de donde pasarían a las neuronas. En estas células la T3 podría actuar directamente en el núcleo o ser un sustrato de D3, al igual que T4. Es posible que en primates el transporte de T4 y T3 dependa exclusivamente de MCT8, por lo que las mutaciones del transportador tienen mayor repercusión que en roedores.")
Hipótesis sobre el transporte y metabolismo de las hormonas tiroideas en el sistema nervioso central (Grijota-Martínez et al67). En la barrera hematoencefálica de roedores el transportador Oatp14 facilita el paso de T4 a los astrocitos, donde se convierte en T3. Mct8 facilita el paso de T4 y T3 probablemente al espacio intersticial, de donde pasarían a las neuronas. En estas células la T3 podría actuar directamente en el núcleo o ser un sustrato de D3, al igual que T4. Es posible que en primates el transporte de T4 y T3 dependa exclusivamente de MCT8, por lo que las mutaciones del transportador tienen mayor repercusión que en roedores.
Varios procesos contribuyen al incremento de T3 circulante (fig. 4): por un lado, un posible aumento de la secreción tiroidea de T3, lo que es aún controvertido63,64, y menor degradación por desyodasa 3 (D3) en órganos como el cerebro. Además, el aumento de T3 circulante incrementa la actividad D1 en el hígado y los riñones. Como consecuencia, se incrementa la conversión de T4 a T3, lo que contribuye a aumentar aún más la T3 circulante. La disminución de T4 circulante se debe a la disminución de la secreción de T4 y al incremento de su conversión a T3 en el hígado y los riñones. Puesto que la desyodasa 2 (D2) se regula de forma negativa por T4, la disminución de ésta produce un incremento de su actividad en el cerebro, incrementándose la conversión de T4 a T3. Esto último consigue compensar, al menos parcialmente, la deficiencia cerebral de T3 en animales experimentales. La disminución de rT3 se explica por el incremento de su degradación por la actividad incrementada de D1 en el hígado y los riñones, y por la menor producción a partir de T4 en el cerebro.

La patogenia del cuadro de afectación neurológica es desconocida y no se tienen datos sobre en qué momento del desarrollo fetal o posnatal comienza a producirse la lesión neurológica ni cuáles son las estirpes celulares afectadas y vías de señalización implicadas. Dado que los ratones knock out para Mct8 no presentan alteraciones neurológicos, no ha sido posible investigar en detalle el síndrome. En ratones existe compensación del hipotiroidismo cerebral, por un mecanismo dependiente de D265, que incrementa el aporte de T3 a partir de la T4 en el cerebro (fig. 3). Los animales no presentan signos de hipotiroidismo cerebral, y sólo algunos de los genes diana de T3 están alterados. En pacientes se ha observado, mediante resonancia magnética, un retraso de la mielinización, que quizás explicaría los déficits neurológicos66. Esta hipótesis se apoya también en la observación de que el cuadro es similar a la enfermedad de Pelizaeus-Merzbacher, como hemos apuntado anteriormente.
Una hipótesis para explicar las diferencias entre ratones y humanos es que en ratones, la presencia de otros transportadores como Oatp1462,67 o Lat-268 en la barrera hematoencefálica permite la paso de T4 al cerebro y la formación de T3 (fig. 3), mientras que en el cerebro humano fetal tanto el paso de T3 como el de T4 dependería en exclusiva de MCT8. Esta hipótesis tiene el apoyo de un estudio muy reciente que demuestra que la barrera hematoencefálica del mono no contiene OATP1C1, el equivalente a Oatp14 de ratón69.
DiagnósticoEn todo paciente con historia de hipotonía neonatal y deficiencia cognitiva se debe medir T4 y T3 total y libre en suero. El incremento de T3, sobre todo si se acompaña de disminución de T4, es casi patognomónico. El diagnóstico se confirma mediante secuenciación del gen.
TratamientoEn la actualidad no existe un tratamiento eficaz aunque se han ensayado algunas medidas terapéuticas. Puesto que los pacientes tienen hipotiroxinemia, se ha ensayado el tratamiento con T452 sin mejoría clínica, e incluso un agravamiento del hipertiroidismo periférico. Para evitar esta última complicación se administró propiltiuracilo para bloquear la función tiroidea, administrando T4 hasta normalizar las concentraciones de T4, T3 y TSH70. No se obtuvo mejoría neurológica, aunque el estado general del paciente mejoró notablemente. En la actualidad se está ensayando una diyodotironina, DITPA (ácido 3,5-diyodo-tiropropiónico). Esta molécula es un agonista del receptor nuclear de T3 al que se une con baja afinidad y posee actividad cardíaca71. De hecho, se encuentra en ensayos clínicos de fase II para el tratamiento de la insuficiencia cardíaca72. DITPA posee la propiedad de pasar la barrera hematoencefálica en ratones, aun en ausencia de Mct8, y poseer una mínima actividad metabólica73. El tratamiento de pacientes con este análogo ha dado buenos resultados en la mejoría del estado general, pero no del neurológico74.
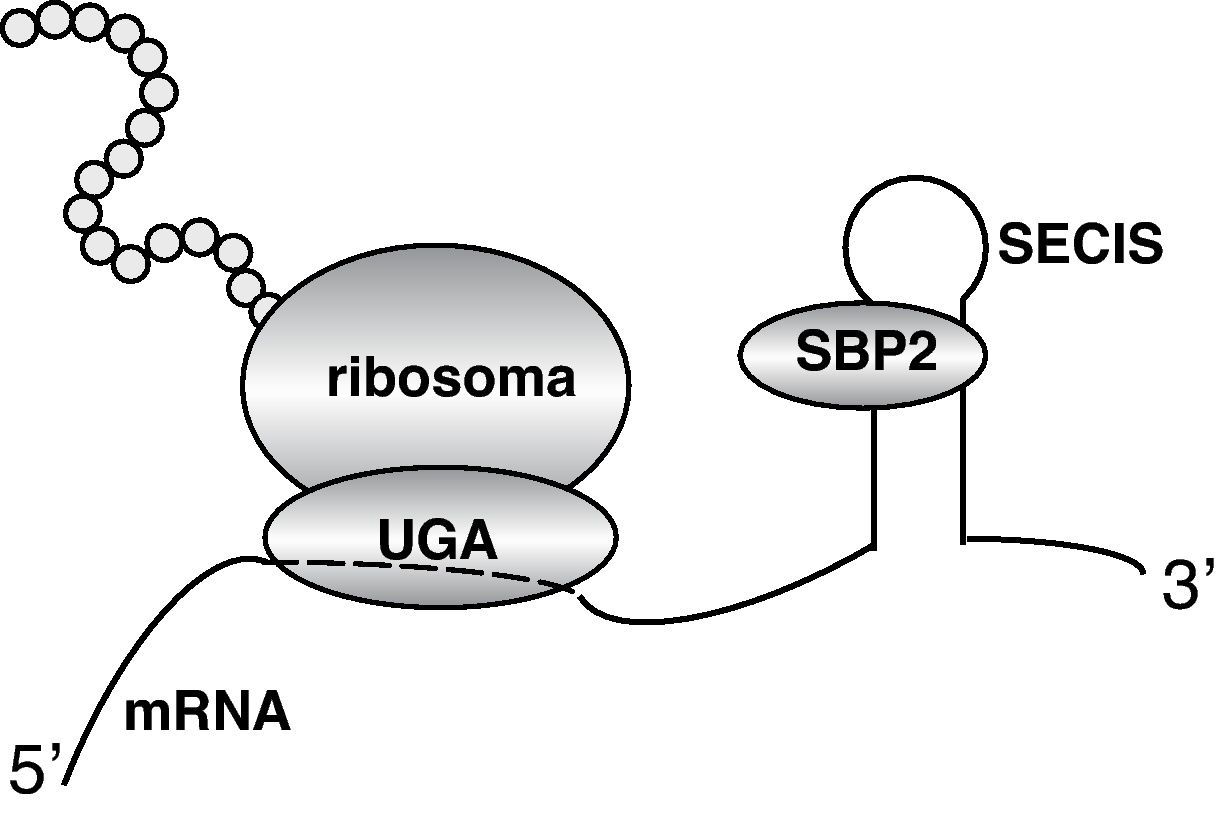
Defecto de desyodaciónEl primer defecto genético del metabolismo de las hormonas tiroideas fue descrito en 2005 por el grupo de Refetoff et al75. En el estudio de la función tiroidea en dos familias con estatura baja y retraso del desarrollo óseo se encontró un perfil de hormonas tiroideas circulantes consistente en T4 y rT3 elevadas y T3 disminuida, justo lo contrario de lo que se ha descrito en la anterior sección sobre mutaciones en MCT8. El estudio bioquímico y molecular usando fibroblastos de los pacientes llevó a la conclusión de que el origen del síndrome era una defecto en las desyodasas causado por mutaciones en la proteína SBP2 (Selenocysteine insertion sequence-binding protein 2 o SECISBP2). Esta proteína es esencial para la síntesis de las desyodasas a nivel de la traducción del mRNA, al igual que de otras selenoproteínas76. El mecanismo de acción de SBP2 consiste en transformar en el mRNA de las selenoproteínas el codón AUG, que normalmente es un codón de terminación, en un codón capaz de reconocer el aminoácido selenocisteína (fig. 5). Para ello, el mRNA de las selenoproteínas posee una secuencia en su extremo 3’, denominada elemento SECIS (Selenocysteine insertion element) al que se une la proteína SBP2. En ausencia de SBP2 funcional, el codón AUG funciona como codón terminador y se produce una proteína truncada sin actividad. La deficiencia de D1 y D2 explica las concentraciones elevadas de T4 y disminuidas de T3, por deficiencia en la conversión de T4 en T3. Las concentraciones aumentadas de rT3 indicarían que la degradación de rT3 por D1 estaría más afectada que la degradación de T4 a rT3 por la D3. El defecto de conversión de T4 en T3 explica que la respuesta de TSH a la administración de T4 en estos pacientes sea defectuosa.

El mRNA de las desyodasas, y en general de todas las selenoproteínas, contienen un codón de terminación UGA que, normalmente no codifica por ningún aminoácido. Sin embargo, en el extremo 3’ del mRNA existe una secuencia denominada elemento SECIS que une la proteína SBP2. Esta proteína interacciona con proteínas del ribosoma haciendo que el codón terminador reconozca el aminoácido Se-cisteína, que se incorpora a la proteína.
Los pacientes, pertenecientes sólo a las dos familias descritas, no presentan una clínica llamativa, aparte de la estatura baja y retraso del desarrollo óseo ya mencionados. Los pacientes son aún jóvenes y no se sabe si con la edad tendrán más predisposición a procesos cancerosos y enfermedades degenerativas, debido a la alteración en otras selenoproteínas implicadas en mecanismos de protección frente al estrés oxidativo.
ConclusiónLos síndromes de resistencia a hormona tiroidea no se limitan en la actualidad a las alteraciones en la acción nuclear de T3, que son las más conocidas. Ante concentraciones de T4, T3 y TSH anómalas no fácilmente explicables, hay que tener presente la posibilidad de alteraciones como las descritas del transporte y de la síntesis de desyodasas, de las cuales aún no se sabe cuál es su incidencia real. En relación con las mutaciones en MCT8, es importante tener en cuenta que los pacientes acudirán en primera instancia a servicios de Neurología pediátrica, en los cuales puede que exista poca familiaridad con la fisiopatología tiroidea. Estos pacientes tenderán a ser diagnosticados como hipotonías congénitas de causa desconocida.
El trabajo en el laboratorio del autor se lleva a cabo con fondos del proyecto integrado CRESCENDO de la Unión Europea, proyectos SAF2008-01168 y SAF2008-00429E del Plan Nacional de I + D + i y el CIBER de Enfermedades Raras, una iniciativa del Instituto de Salud Carlos III.
Siguiendo las normas de National Center for Biotechnology Information (NCBI), el acrónimo del gen o proteína humanos se representa con todas las letras en mayúsculas, mientras que los de roedores se representan con la primera letra en mayúscula y las siguientes en minúsculas. En ambos casos, el nombre del gen se representa en cursiva.
