


En 2006, el Consenso de Chicago1 sobre el manejo de los trastornos intersexuales propuso la sustitución de términos como intersexuales, hermafroditas y pseudo-hermafroditismo por el término «trastornos del desarrollo sexual», definido como enfermedades congénitas en los que el desarrollo de los cromosomas, las gónadas, o sexo anatómico es atípico. El nacimiento de un niño con alguna de estas características tiene indiscutiblemente un gran impacto social y psicológico para sus familiares, por lo tanto, es necesaria una rápida evaluación a nivel clínico, hormonal, genético, molecular y radiológico que permita determinar la etiología del caso, así como la orientación terapéutica. La evaluación inicial del niño con ambigüedad genital debería incluir: historia familiar, examen físico completo, evaluación de los cromosomas sexuales y un estudio de la anatomía interna, así como los niveles de la secreción esteroidea gonadal y adrenal. Sin embargo, el diagnóstico de estos niños con sexo no diferenciado es complejo, debido a una etiopatogénesis multifactorial y muy variable.
Se trata de una mujer de 19 años de origen rumana, diagnosticada de hiperplasia suprarrenal congénita en tratamiento farmacológico, derivada a la consulta de Genética Clínica desde el Servicio de Endocrinología. Como antecedentes personales, extirpación de hipertrofia del clítoris a los 3 años previo estudio por laparotomía que constataba la presencia de útero y ovarios. No presenta ningún antecedente familiar de interés. Refería amenorrea primaria e hirsutismo. En el examen físico general se apreció una estatura de 1,50m, peso de 105kg, obesidad troncular, fenotipo femenino y signos de virilización intenso en cara, brazos, tórax y piernas, voz grave, genitales externos femeninos normales.
Excepto un nivel bajo del estradiol (<10pg/mL), en el resto de los estudios bioquímicos realizados se obtuvieron niveles entre los límites normales, incluida la 17 hidroxiprogesterona (17OHP) de 0,91ng/mL. La ecografía ginecológica presentaba un útero en anteversión regular y vacío con una histerometría de aproximadamente 59×17×30mm, atrófico para la edad de la paciente, e imágenes hiperecogénicas ovoideas de 19×7mm y 29×16mm en el lado derecho e izquierdo, respectivamente, sugestivas igualmente de ovarios atróficos para la edad de la paciente. La resonancia magnética (RM) de abdomen confirmó la presencia de útero y ovarios marcadamente atróficos sin otras anormalidades genitourinarias. Otra RM constató que las glándulas suprarrenales se encontraban dentro de la normalidad.
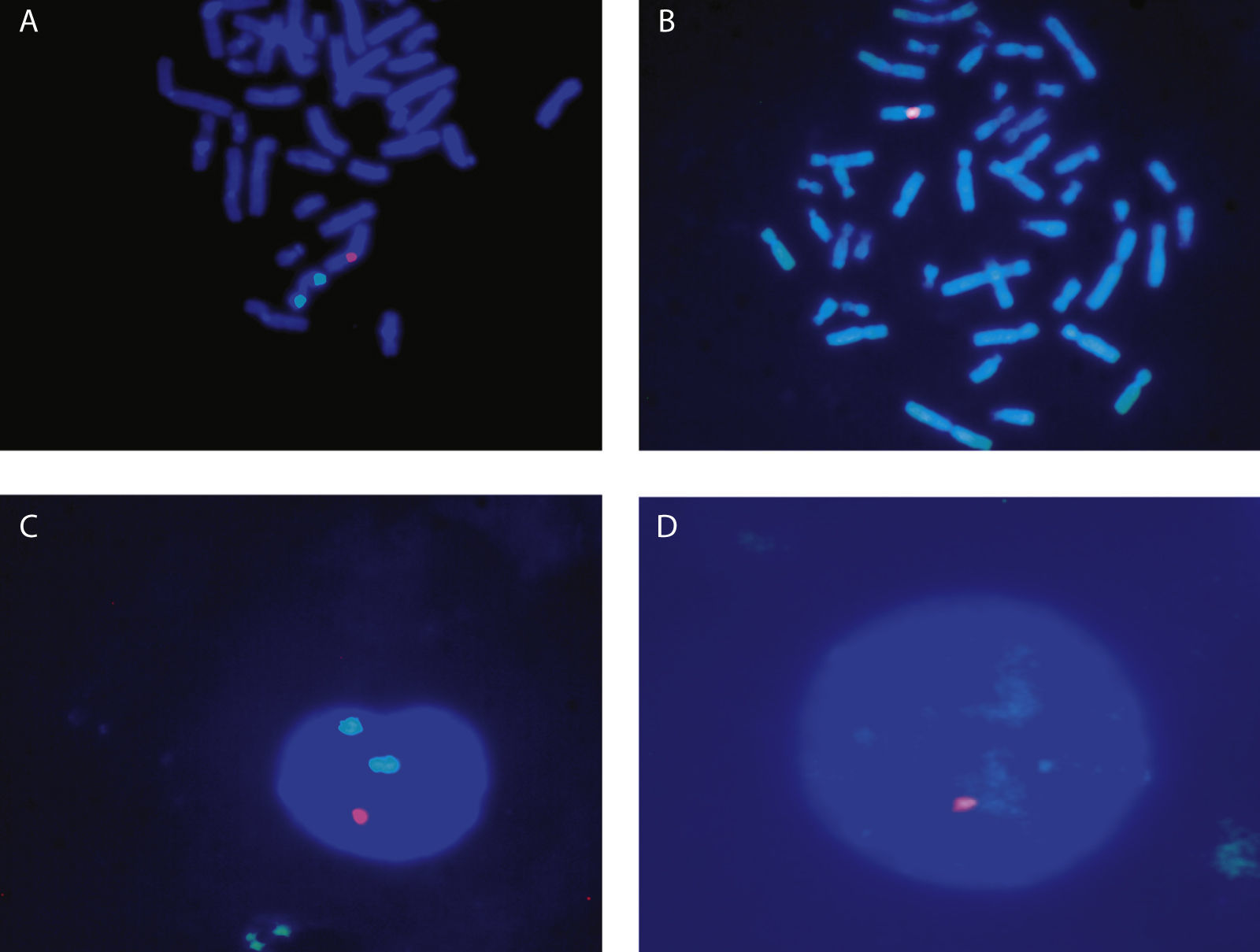
El estudio molecular de la hiperplasia suprarrenal congénita no presentó ninguna mutación responsable del desarrollo de la enfermedad. Se realizó el estudio del cariotipo en sangre periférica que presentó: en 14 metafases analizadas 46 cromosomas con fórmula sexual XY y en 6 metafases estudiadas tenían 45 cromosomas con fórmula sexual X. Se realizó la técnica de citogenética molecular de hibridación in situ fluorescente (FISH) con sonda centromérica del cromosoma X e Y [Vysis CEPX (DXZ1)/Y(DYZ1)]. La sonda DXZ1 mostró una señal para la región centromérica del cromosoma X en el 30% de los núcleos mientras que la sonda DYZ1 manifestó dos señales para la región centromérica del cromosoma Y en el 70% de los núcleos (fig. 1). La fórmula cromosómica convencional resultó: mos 45, X[30] ish (cepX[DXZ1]+)/46, X, dic(Y) [70] ish (cepX[DXZ1]+/Y[DYZ1]++). No se pudo realizar el estudio familiar porque no vivían en España (fig. 1).
 y el cromosoma Y (verde). A y B: metafases, C y D: núcleos. A: metafase con 2 señales verdes (2 centómeros de cromosoma Y) y una señal roja (un centrómero del cromosoma X), B: metafase con una señal roja (un centrómero del cromosoma X) C: núcleo con 2 señales verdes (2 centrómeros del cromosoma Y) y una señal roja (un centrómero del cromosoma X), D: metafase con una señal roja (un centrómero del cromosoma X).")
FISH realizada con sonda centromérica para el cromosoma X (rojo) y el cromosoma Y (verde). A y B: metafases, C y D: núcleos. A: metafase con 2 señales verdes (2 centómeros de cromosoma Y) y una señal roja (un centrómero del cromosoma X), B: metafase con una señal roja (un centrómero del cromosoma X) C: núcleo con 2 señales verdes (2 centrómeros del cromosoma Y) y una señal roja (un centrómero del cromosoma X), D: metafase con una señal roja (un centrómero del cromosoma X).
El estudio de la región determinante del sexo (SRY), de la región del factor de azoospermia (AZF) y del gen de la supresión de azoospermia (DAZ) mediante la reacción en cadena de la polimerasa (PCR), utilizando sondas de 20 locis a los largo del cromosoma Y (SRY,DYS271, DYS148, DYS273, KALY, DYS212, SMCY, DYS215, DYS218, DYS219, DYS221, DYS223, DYS224, DYF51S1, DYS236, DAZ, DYS240), detectó la presencia de todas las regiones estudiadas del cromosoma en las células de sangre periférica del paciente
El diagnóstico definitivo fue síndrome de Turner, mosaicismo 45,X/46,X, dic (Y).
La hiperplasia suprarrenal congénita se debe a una deficiencia en una u otra de las enzimas de la biosíntesis de cortisol. En aproximadamente el 95% de los casos, se debe a la deficiencia de la 21-hidroxilasa que se altera en zona fascicular de la corteza suprarrenal de tal forma que la 17OHP no se convierte a 11 desoxicortisol. Debido a la síntesis defectuosa de cortisol, se incrementan los niveles de ACTH, lo que resulta en la sobreproducción y acumulación de precursores de cortisol, en particular 17OHP. Esto hace que la producción excesiva de andrógenos produzca virilización2. El caso presentado estaba en tratamiento por una hiperplasia suprarrenal congénita y con un antecedente personal de intervención de hipertrofia del clítoris. Con el estudio molecular realizado se descartó que la paciente presentara una mutación causante del trastorno del desarrollo sexual.
El mosaicismo de los cromosomas sexuales, que consiste en un 45,X asociado con otra línea celular que contiene un cromosoma Y estructuralmente normal o anormal, representa una de las principales causas de genitales ambiguos3. En la mayoría de los casos de mosaicismo 45,X/46,XY, la causa se considera la pérdida de la no disyunción del cromosoma Y después de la fertilización normal disómica4. Se estima un 7% la frecuencia del cromosoma o fragmentos de cromosoma Y en el síndrome de Turner5. La proporción del mosaicismo y los tejidos afectados determina la expresión clínica del defecto. En un porcentaje elevado de pacientes en el que el cariotipo presenta la línea 45,X, aparecen estigmas turnerianos de intensidad muy variable como es el caso de nuestra paciente. Después de la pubertad se suele producir un aumento de gonadotropinas y una producción de andrógenos muy variable de unos pacientes a otros que determina una virilización de intensidad también muy variable.
El gen de la región determinante del sexo (SRY del cromosoma Y) es un factor de transcripción en el brazo corto del cromosoma Y que es crítico para el desarrollo de los testículos. Las mutaciones en el gen de SRY en los individuos XY conlleva un desarrollo fenotípico femenino, mientras que los pacientes con cariotipo 45,X portador del gen SRY en un autosoma tienen un fenotipo masculino6. La temprana detección de secuencias del cromosoma Y es importante debido al alto riesgo de desarrollo de tumores gonadales especialmente gonadoblastoma, que es una rara neoplasia gonadal. La mayoría de los ellos se desarrolla en mujeres portadoras de un cariotipo anormal en que al menos una parte de la región centromérica del brazo corto del cromosoma Y está presente, una región que a menudo se refiere como el locus GBY7. Se estima el riesgo de gonadoblastoma o disgerminoma aproximadamente entre un 10%5 y un 35%8. Ante el mosaicismo de cromosoma X y el cromosoma Y, y la detección de secuencias específicas del cromosoma Y en los pacientes con mosaicismo 45X/46XY es necesaria con el fin detectar el riesgo de gonadoblastoma y de recomendar la gonadodectomía.
Pacientes diagnosticados de trastorno en el desarrollo sexual pueden tener alteraciones psicosociales. Nuestra paciente se consideraba mujer, y el hecho de que conocía la línea celular conteniendo el cromosoma Y no le generó ningún conflicto psicológico. La identidad de género es un tema complejo y sensible, y el médico debe tener un debate abierto y franco con la persona y en el caso que sea necesario se debe plantear la derivación a profesionales psiquiatras o psicólogos expertos que atiendan trastornos de identidad sexual.
A pesar de haber recibido consejo genético adecuado, la paciente no quiso realizar la gonadectomía preventiva y exéresis de estructuras wolffianas para evitar el riesgo de tumores y porque puede producir virilización.
En resumen, a la paciente, a los 3 años, se le realizó una laparoscopia confirmando útero y ovarios y se intervino de hipertrofia del clítoris. La paciente fue diagnosticada y tratada como una hiperplasia suprarrenal congénita. A los 19 años se le realizó un cariotipo en sangre periférica presentando dos líneas celulares, 45,X/46,XY, siendo el cromosoma Y dicéntrico, es decir, un cromosoma constituido por dos segmentos que provienen de dos cromátides de un mismo cromosoma9. Dado que el cariotipo no siempre identifica el cromosoma o fragmentos del cromosoma Y, se debe de realizar estudios moleculares o de FISH utilizando una sonda centromérica Y, ante cualquier paciente con síndrome de Turner con un cromosoma marcador o un fragmento de sexo cromosómico de origen desconocido, y complementar el estudio si fuera necesario10. El desarrollo de las técnicas moleculares y citogenéticas durante los últimos años han permitido confirmar y reconducir el diagnóstico y el tratamiento en este caso presentado.
