


En los últimos años se ha empezado a descubrir las bases genéticas de la carcinogénesis en diferentes neoplasias humanas. La desregulación de la vía de señalización dependiente de la proteincinasa de activación mitogénica (MAPK) es un mecanismo fundamental en la oncogénesis del carcinoma papilar de tiroides (CPT), el tumor endocrino más frecuente. Concretamente, tres integrantes de la vía –el receptor tirosincinasa RET, Ras y BRAF– se encuentran implicados en más del 70% de los casos. El reordenamiento cromosómico de RET, denominado RET/PTC, ocasiona la activación enzimática de esta tirosincinasa. Fue la primera alteración genética específica del CPT y se encuentra en un 5-70% de las muestras tumorales. En menor proporción se han encontrado activaciones constitutivas de otros receptores tirosincinasa, como NTRK1, c-met o EGFR. La mutación de BRAF es la alteración genética más frecuente en el CPT. En la mayoría de los casos, la mutación ocasiona un cambio de valina por ácido glutámico en la posición 600 (V600E). Finalmente, Ras es la molécula menos afectada de la vía. La presencia de estas alteraciones se correlaciona, en algunos casos, con el comportamiento clínico de la enfermedad. Así, la mutación de BRAF condiciona una mayor agresividad y un peor pronóstico. No hay tanto consenso en el papel de RET/PTC. Por otro lado, el descubrimiento de estas alteraciones abre la puerta a nuevas estrategias terapéuticas, en especial para los pacientes en quienes el tratamiento convencional resulta inefectivo. Entre las nuevas opciones se encuentran los inhibidores de las tirosincinasas. Algunos fármacos de esta familia han empezado a ser utilizados con resultados esperanzadores.
In recent years, significant progress has been made in elucidating the genetic bases promoting tumorigenesis in various human neoplasms. Constitutive activation of the mitogen-activated protein kinase (MAPK) signaling pathway is a major event in the carcinogenesis of papillary thyroid carcinoma (PTC), the most prevalent endocrine malignancy. Affected elements include RET/PTC rearrangements and point mutations of the Ras and BRAF genes. Mutations in these genes are found in over 70% of PTC. Chromosomal RET rearrangements, called RET/PTC, result in constitutive ligand-independent activation of RET kinase, which was the first genetic anomaly detected in PTC and is found in 5-70% of tumoral samples. Although less frequent, the activation of other tyrosine kinase receptors, such as NTRK1, c-Met or EGFR, has also been reported in PTC. The BRAF mutation represents the most common genetic alteration found in PTC. More than 90% of BRAF mutations lead to a change of a valine to a glutamic acid at position 600 (V600E). Finally, Ras is the least affected molecule in the pathway. A relationship between clinical behavior and these genetic alterations has been proposed. Thus, the BRAF mutation is associated with a more aggressive PTC phenotype and is correlated with poorer outcomes. However, no clear association has been found between RET/PTC and clinical features. The discovery of these alterations opens the way to new therapeutic strategies, especially to treat those patients in whom conventional therapy is not effective. Several new drugs are being tested, such as small molecule tyrosine kinase inhibitors. Some of these recently developed agents have begun to be used with promising results.
El cáncer de tiroides es la neoplasia endocrina más frecuente. En un 3% de los casos afecta a las células C o parafoliculares y da lugar al carcinoma medular. Otro 2% de los casos corresponde a la forma indiferenciada denominada carcinoma anaplásico. Sin embargo, la gran mayoría se engloba en el término carcinoma diferenciado de tiroides (CDT), con dos formas principales, el folicular (15%) y el papilar (CPT) (80%)1. El CPT incluye un grupo heterogéneo de neoplasias tiroideas compuesto por diversas variantes con implicaciones para el pronóstico también diferentes. Actualmente, el esquema terapéutico comprende la tiroidectomía total, con exéresis simultánea de los ganglios del compartimento central, la ablación de restos con radioyodo (131INa) y el tratamiento con L-tiroxina a dosis supresoras de tirotropina. Mediante este protocolo, se ha logrado una gran mejora del pronóstico de la enfermedad, con una mortalidad que no excede el 10% a los 10 años2, 3. Por esta razón, algunos investigadores clínicos han venido considerando que la modalidad terapéutica podría ser demasiado agresiva para la mayoría de los casos. Así, y a fin de seleccionar qué pacientes pudieran tener una evolución peor, se han propuesto varios factores pronósticos. Los más aceptados son la edad, el tipo histológico, la extensión inicial de la enfermedad y el tamaño del tumor primario. A pesar de ello, no se ha establecido la causa de que en algunos pacientes el CPT sufre un proceso de desdiferenciación que hace que el tejido neoplásico pierda la capacidad de producir tiroglobulina y concentrar yodo, lo que convierte el tumor en resistente al tratamiento convencional.
El conocimiento cada vez más profundo de las bases moleculares que rigen la transformación neoplásica ha puesto de manifiesto que la manera de caracterizar correctamente el cáncer de tiroides debe residir en los factores genéticos involucrados en los mecanismos que determinan la extensión inicial de la enfermedad y el proceso de desdiferenciación.
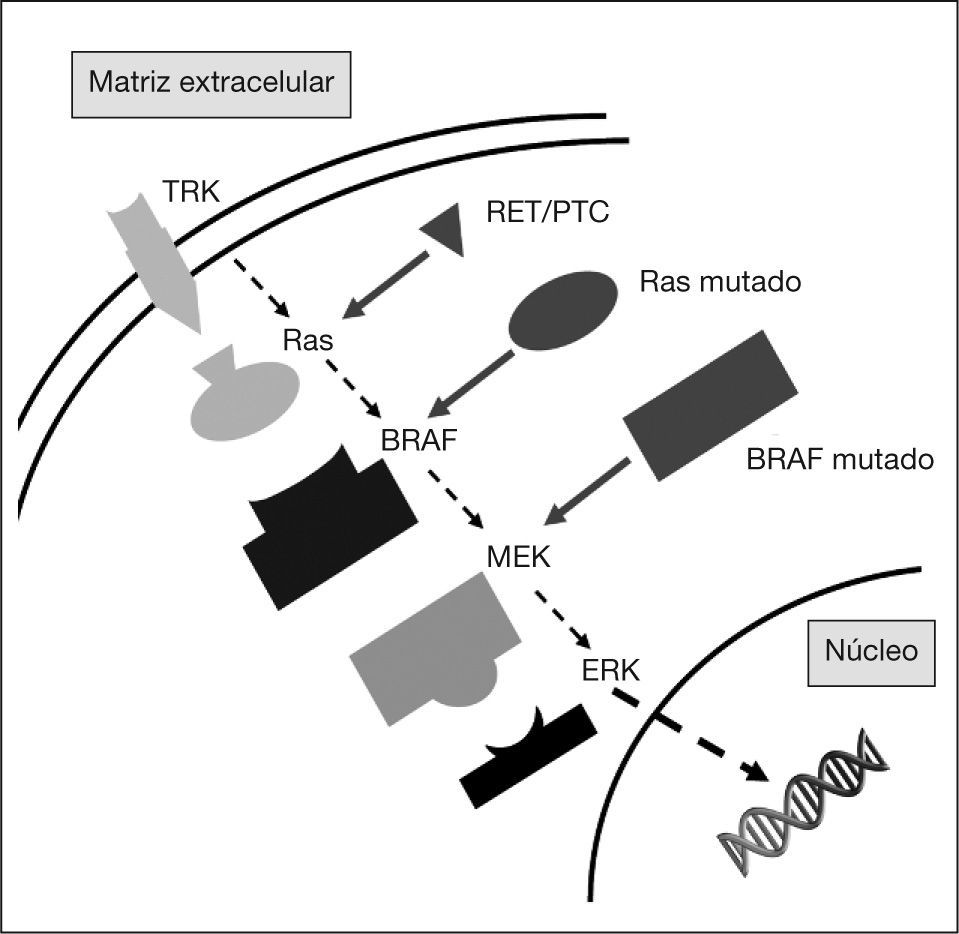
Hace poco más de 20 años Sturgill et al4 detectaron la presencia de actividad proteincinasa activada por insulina en extractos de adipocitos 3T3-L1 con capacidad para fosforilar un péptido identificado como MAP- 2. Durante estas dos décadas se ha llegado a detallar, con elevada exactitud, la composición de los elementos que integran lo que hoy conocemos como vía de señalización dependiente de la proteincinasa, mitogen-activated protein kinase (MAPK). Esta ruta, altamente conservada desde microorganismos hasta mamíferos, constituye una de las cascadas moleculares más importantes en la regulación de aspectos clave de la funcionalidad celular como la diferenciación, la proliferación y la migración5. Su activación transmite señales desde distintos tipos de receptores de membrana hasta activar factores de transcripción nuclear (fig. 1). Una pequeña proteína de unión a guanosintrifosfato (GTP), denominada ras, es la molécula que actúa como transductor inicial y común de la señal iniciada tras la activación de los receptores de membrana. Ras se comporta como un interruptor bimodal, con dos estados conformacionales, uno activo y otro inactivo. La conformación activa produce la activación de la MAP-cinasa-cinasa-cinasa, conocida también como RAF. A su vez, RAF activa, mediante fosforilación, la MAP-cinasa-cinasa o MEK. Finalmente, MEK activa la MAP-cinasa o ERK. Por lo tanto, la estructura básica de MAPK está formada por un módulo de tres cinasas (RAF, MEK y ERK) dependiente de la proteincinasaque se activan de manera secuencial. Mientras que RAF y MEK son de localización citoplásmica, ERK tiene la capacidad de traslocarse al núcleo, y ahí puede fosforilar directamente un gran número de factores de transcripción como c-jun o c-Myc.
. A la derecha: lugar de activación constitutiva de la vía debido a las diferentes alteraciones genéticas detectadas en el carcinoma papilar de tiroides")
En los últimos años se ha visto que la desregulación de MAPK está implicada en la carcinogénesis de numerosos tumores humanos. De hecho, una gran cantidad de miembros de la vía fueron identificados inicialmente como protooncogenes. Así, la activación aberrante de la vía MAPK es un hallazgo frecuente en células malignizadas. En la última década, se ha podido demostrar que la alteración de la vía constituye un elemento precoz, y necesario, en el desarrollo del CPT6. Concretamente, tres elementos de la vía ya han sido implicados en más del 70% de los casos, y se acepta que la desregulación de MAPK es un evento inicial en la carcinogénesis específica de este tipo de tumor tiroideo7. Estos elementos son el receptor tirosincinasa RET, la proteína Ras y la cinasa RAF. Dichas alteraciones son mutuamente excluyentes8, lo que refuerza la idea de que la aberración de la vía de la cual forman parte es un elemento indispensable para el desarrollo del CPT9. Así, en un elegante conjunto de estudios, Melillo et al10 han demostrado que las tres moléculas están implicadas en la iniciación del CPT a través de la misma cascada de señalización.
RECEPTORES TIROSINCINASALa vía MAPK puede ser activada a partir de diversas señales extracelulares, como citocinas, factores de crecimiento y otros péptidos, a través de su acción sobre receptores de membrana. Entre las diferentes familias de receptores capaces de activar ras se encuentran los receptores con actividad tirosincinasa (RTK). Todos los integrantes de este grupo presentan una estructura común formada por tres regiones: un dominio extracelular que constituye el lugar de unión con el ligando, el dominio transmembrana y, en tercer lugar, el dominio intracelular o citoplásmico, que es el que posee la actividad enzimática tirosincinasa. En las mismas fechas que Sturgill y Ray sentaban las bases sobre el descubrimiento de la vía MAPK, Fusco et al11 describían la presencia de una aberración cromosómica en el gen de un RTK, conocido como RET, en el CPT.
RET/PTCEl gen RET (rearranged during transfection)12 codifica un RTK que se expresa primariamente en neuronas sensoriales del sistema simpático y del plexo entérico, así como en la yema del uréter durante la embriogénesis y en la diferenciación de los espermatogonios13. El ligando natural del receptor es el factor de crecimiento GDNF (glial line-derived neurotrophic factor). RET está implicado en diferentes afecciones humanas. Así, la mutación inactivadora germinal causa la agangliosis del tracto intestinal (enfermedad de Hirschprung), mientras que mutaciones activadoras son las causantes de la neoplasia endocrina múltiple tipo 2A (MEN 2A)14.
Fusco et al11 describieron en 1987 la presencia de una recombinación pericentromérica del cromosoma 10, por la cual se producía una fusión del dominio tirosincinasa de RET con el extremo 5' terminal de un gen conocido como H4. Este tipo de aberración estructural cromosómica es un mecanismo común en la oncogénesis de las neoplasias hematológicas, pero es un hallazgo muy poco frecuente en los carcinomas15. El gen quimérico resultante fue denominado RET/PTC. El efecto de dicha alteración es la activación enzimática constitutiva de RET. Posteriormente han sido descritas otras variantes de RET/PTC, en función del gen que se fusiona con el dominio tirosincinasa (TK) de RET y que actúa de promotor en su expresión. Hasta la fecha se conocen más de 17 isoformas, de las que RET/PTC1 y RET/PTC3 son las encontradas más frecuentemente16-17.
En la actualidad no hay ninguna duda de que RET/ PTC es un elemento clave en la carcinogénesis del CPT. Estudios experimentales han demostrado que induce la transformación neoplásica en cultivos de células foliculares18. Además, ratones transgénicos portadores de RET/PTC desarrollan el tumor con una elevada frecuencia19. También se acepta que su papel etiopatogénico queda restringido específicamente al tipo histológico papilar, ya que no ha sido detectado en otras clases de neoplasias tiroideas, como el carcinoma folicular o el carcinoma medular17. No obstante, algunos autores han detectado RET/PTC en muestras de tiroides con tiroiditis crónica autoinmunitaria, aunque se desconoce el significado exacto de este hallazgo20.
RET/PTC se encuentra frecuentemente en microcarcinomas papilares21. Por otro lado la presencia de RET/ PTC es suficiente para ocasionar cambios morfológicos característicos de CPT en el núcleo de células foliculares22. Además, RET/PTC es más predominante en las formas de CPT bien diferenciadas, como la variante clásica23, que en variantes histológicas más agresivas, como el carcinoma pobremente diferenciado y el carcinoma anaplásico24. Todo ello lleva a considerar que RET/PTC actúa en los primeros pasos de la transformación tumoral, pero no participa en los eventos posteriores25.
El factor etiológico que provoca RET/PTC es desconocido. Se ha publicado su elevada presencia en los casos de CPT aparecidos tras el accidente de la central nuclear de Chernobyl26. Además, estudios experimentales han mostrado que la irradiación con rayos X induce la formación de RET/PTC en cultivos de células foliculares27, 28. Recientemente, Hamatani et al29 han detectado una elevada incidencia de RET/PTC en casos de CPT de supervivientes de la bomba atómica. Todo ello parece indicar que las radiaciones ionizantes podrían ser la causa de la aberración cromosómica. Nikiforova et al30 han señalado que la cromatina de las células tiroideas presenta una estructura tridimensional específica que la convertiría en altamente sensible a esta anomalía genética. Por otro lado, se ha visto que las células tiroideas son relativamente resistentes a la apoptosis ante la lesión de su material genético31. Así, la combinación de una configuración tridimensional característica y la disminuida respuesta a la apoptosis justifican la especial predisposición a la aberración cromosómica32.
Desde su descubrimiento, son numerosos los trabajos que han analizado la incidencia RET/PTC en el CPT, pero se observa gran disparidad de resultados. Así, la frecuencia oscila desde el 2,5 hasta más del 80% según las series estudiadas33-38. La heterogeneidad detectada ha sido atribuida a diferentes causas, como factores geográficos, características de los pacientes que configuran las distintas series, variantes histológicas o la metodología empleada34, 39, 40. Por ejemplo, las series que incluyen a pacientes más jóvenes encuentran mayor incidencia41. El método utilizado para su determinación podría ser de lo más relevante42. Inicialmente, la detección de RET/PTC se efectuaba mediante técnica molecular y posteriormente se dispuso de immunohistoquímica43. Ésta permite la detección de cualquier variante de la alteración, mientras que la primera es específica para cada una de ellas36. Por ello los trabajos que utilizan determinaciones immunohistoquímicas hallan una mayor frecuencia de casos positivos que supera, en muchas ocasiones, el 60% de las muestras analizadas37, 44. Finalmente, algunos autores han descrito heterogeneidad en la distribución de RET/PTC en las células de un mismo tumor45.
Otro aspecto controvertido es la utilidad clínica que puede ofrecer RET/PTC. No hay un claro consenso sobre el valor pronóstico de esta alteración. Algunos investigadores postulan que la presencia de RET/PTC se asocia a tumores con bajo potencial de crecimiento, a pacientes más jóvenes, a los que exhiben características histológicas de la variante clásica e incluso a neoplasias de menor tamaño, como es el caso del microcarcinoma24, 35, 36, 46. De esta manera, numerosos autores no han encontrado ninguna relación entre RET/PTC y los factores pronósticos clásicamente aceptados como la edad del paciente, el tamaño del tumor o la extensión36, 47-49. Tampoco se han observado diferencias significativas en la supervivencia entre los pacientes RET/ PTC positivos y los negativos47. Sin embargo, otros autores han demostrado que RET/PTC se asocia de manera significativa a una mayor incidencia de extensión extraglandular, pero limitada especialmente a la invasión de ganglios linfáticos regionales37, 50, 51. A pesar de esta asociación, hay cierta unanimidad en que los portadores de RET/PTC tienen bajo potencial de malignidad, aunque se desconoce la causa de esta aparente paradoja46.
En resumen, RET/PTC fue la primera alteración genética específica descrita en el CPT. Se lo considera un evento precoz en el proceso de carcinogénesis de este tipo de neoplasia y su presencia define un patrón histológico de bajo grado de malignidad, aunque puede asociarse a una mayor predisposición de afección ganglionar cervical.
NTRK1El papel de la activación de enzimas con actividad TK como paso inicial en el proceso de transformación neoplásica en el CPT se vio reforzado en el momento en que se describió la implicación de un segundo RTK. NTRK1 (neurotrophic tyrosine kinase receptor type 1) es el receptor del NGF (nerve growth factor), el cual regula el crecimiento y la diferenciación de neuronas tanto del sistema nervioso central como del sistema nervioso periférico52. La mutación con pérdida de función del gen ha sido implicada en la enfermedad genética conocida como insensibilidad congénita al dolor con anhidrosis53. En el CPT, la alteración detectada es parecida a la de RET, es decir, un reordenamiento cromosómico con la fusión del dominio enzimático con otros genes, que actúan de promotores activando su función TK54. Hasta la fecha se han descrito cuatro variantes en relación al gen fusionado55. La implicación de NTRK1 en la génesis del CPT ha sido confirmada experimentalmente por Russell et al56. Los autores desarrollaron ratones transgénicos que expresaban TRKT1 (la variante más frecuente, fruto de la fusión del dominio enzimático de NTRK1 con el gen TPR) y observaron que todos los animales mayores de 7 meses de edad desarrollaban hiperplasia y/o carcinoma tiroideo. Por otro lado, Roccato et al57 han demostrado que la estructura tridimensional del cromosoma 1, donde se localiza el gen, favorece la fusión de NTRK1 con TPR. No obstante, y a diferencia de lo que ocurre con RET, no parece que las radiaciones ionizantes tengan un papel etiológico en el reordenamiento de NTRK155. Así, la incidencia en casos de CPT tras Chernobyl es baja y no muestra diferencias de la encontrada en CPT esporádicos58, 59.
La incidencia de NTRK en el CPT es menor que la de RET/PTC, aunque presenta también una gran variación que oscila, según las series, entre 0 y el 50% de las muestras analizadas49, 60, 61. Algunos autores han determinado conjuntamente la presencia de RET/PTC y NTRK1. En todos los casos la incidencia del primero es superior a la del segundo, y la suma de ambos alcanza alrededor del 30% de las muestras49, 60. Cabe destacar que no existe ningún caso publicado en que coexistieran ambas alteraciones en un mismo tumor60, 62.
El significado clínico de NTRK1 no está establecido. Algunos autores han encontrado que los portadores de esta alteración tienen peor pronóstico que los pacientes con RET/PTC49, pero el hallazgo no ha sido verificado por otros estudios60.
EGFRAparte de RET y NTRK1, otros RTK han sido implicados en la carcinogénesis del CPT, aunque en muchos casos los estudios han mostrado resultados contradictorios. Así, algunos trabajos han encontrado una sobreexpresión del receptor del factor de crecimiento epidérmico (EGFR)63. Además, para ciertos autores la presencia de dicho receptor se asocia a tumores de comportamiento más agresivo64, 65. En otras series se ha observado una diferencia de localización celular de EGFR, siendo característica su ausencia en el núcleo de las células del CPT, contrariamente a lo que ocurre en el carcinoma folicular66. Finalmente, trabajos más recientes apuntan que EGFR podría tener un papel más importante en los tumores anaplásicos o pobremente diferenciados67, 68. En este sentido, Liu et al69 han resaltado la elevada implicación de diversos TRK en los carcinomas más indiferenciados.
C-METLa participación de c-met, el receptor del factor de crecimiento de los hepatocitos (HGF), ha sido ampliamente estudiada en el CPT. La mayoría de los trabajos coinciden en encontrar una sobreexpresión del gen que, en algunas series, llega a superar el 90% de las muestras analizadas70, 71. No obstante, no hay tanta unanimidad en cuanto a su posible significado. Para algunos autores la presencia de c-met se asocia a un mayor grado de extensión y con ello a un peor pronóstico70, 72, 73. Una posible causa de esta asociación puede ser que la sobreexpresión de c-met es más frecuente en variedades histológicas más agresivas74, 75. A pesar de ello, en otros estudios c-met carece de significado clínico76, 77.
RASEl protooncogén Ras codifica una proteína de 21 kDa que actúa como transductor de señales de receptores de factores de crecimiento. Las mutaciones del dominio del GTP (codón 12–13) o de la GTPasa (codón 61) producen un cambio en la secuencia de aminoácidos cuyo resultado es su activación constitutiva. Entre sus muchos efectos reguladores, cabe destacar la acción inhibidora sobre p27, proteína de la familia de las CIP/KIP que regula el ciclo celular. Así, el efecto neto de Ras es una estimulación en la progresión del ciclo de la célula78.
La mutación de Ras es uno de los trastornos más prevalentes, ya que afecta a un 30% de los cánceres en la especie humana79. Se han descrito mutaciones en los tres genes de Ras: H-Ras, K-Ras y N-Ras. Todas ellas fueron las primeras en ser asociadas a carcinomas tiroideos y han sido encontradas en las diversas variedades histológicas del cáncer de dicha glándula. De igual forma que los reordenamientos de PAX8-PPAR7, son mucho más frecuentes en el carcinoma folicular (CFT) y en la variante folicular del CPT, así como en neoplasias tiroideas benignas80-83, mientras que son muy poco frecuentes en el CPT84. No hay diferencias entre el tipo de mutación y el desarrollo de las distintas variantes o la evolución clínica. Como en el caso de RET/PTC, se considera que es una alteración propia de las etapas tempranas de la carcinogénesis85, 86. Los estudios realizados con líneas celulares de neoplasias epiteliales indican que es un potente inhibidor de la apoptosis87, 88. Se ha observado que en ausencia de tirotropina la expresión de H-RASV12 acelera la proliferación celular, lo cual induce el crecimiento tumoral no dependiente de tirotropina y afecta, principalmente, a las células que, debido al proceso de desdiferenciación mencionado anteriormente, han perdido la capacidad de respuesta a la tirotropina89. Este fenómeno ocurre en los casos con evolución desfavorable. De hecho, se ha visto que, cuando la expresión de Ras es muy elevada, se produce una inhibición de los genes específicos del tiroides como TTF1 y PAX8, dos factores de trascripción necesarios para mantener la diferenciación de las células foliculares tiroideas90. Este hecho ha sido corroborado en otros trabajos, en los que se ha hallado una decreciente expresión de factores que indican la diferenciación de los tirocitos, como la tiroperoxidasa91. También se han relacionado las mutaciones de Ras con lesiones del ADN, como alteraciones en el alineamiento de los cromosomas durante la mitosis, formación de micronúcleos y amplificación de los centrosomas92.
La presencia de mutaciones de Ras en neoplasias benignas de tiroides y su mayor incidencia en el carcinoma folicular y en las variantes foliculares del CPT han limitado su uso como factor pronóstico. Sin embargo, algunos autores han encontrado que la mutación del gen se asocia a estadios más avanzados del CPT93, 94. Otros trabajos han hallado una relación entre la mutación de Ras y la evolución a carcinomas poco diferenciados o anaplásicos y señalan que estos casos deberían ser controlados de una forma más estrecha95. Así pues, se trata de una mutación muy poco frecuente en el CPT, y más característica de los carcinomas poco diferenciados. En estos casos podría constituir un factor de mal pronóstico96, 97.
BRAFLos genes RAF fueron identificados inicialmente como oncogenes en tumores que afectaban a ratones y aves98. Se han identificado tres genes, llamados A-RAF, B-RAF y RAF-1 (o C-RAF). En las células foliculares del tiroides, la isoforma B-RAF es la predominante6. Como hemos visto, Ras se encuentra en la membrana y puede ser activado por factores de crecimiento, hormonas y citocinas99. La activación de esta vía produce la fosforilación de BRAF que activa, constitutivamente, ERK e inicia así un gran número mecanismos celulares implicados en la carcinogénesis100. Se han detectado más de 40 mutaciones oncogénicas de estos genes en un gran número de cánceres humanos. Destacan, entre otros, el melanoma, el adenocarcinoma de colon, el carcinoma de ovario y el CPT101. En este tumor la mutación de BRAF más frecuente es la V600E (anteriormente llamada V599E) que, a causa de un cambio de timidina por adenosina en el nucleótido 1796, hace que la posición sea ocupada por ácido glutámico en lugar de valina102. Este cambio produce una alteración en la conformación de la molécula de B-RAF que la convierte en una estructura activada103. Así, la forma mutada muestra una actividad enzimática unas 500 veces superior que la molécula no mutada104.
La mutación V600E de BRAF es específica del CPT y su prevalencia oscila entre el 35 y el 40% en la mayoría de los estudios9, 105-108, aunque en algunas series supera el 60%109. También se ha relacionado con carcinomas anaplásicos que se originan a partir de la desdiferenciación de un CPT110. No se ha observado en carcinomas foliculares, medulares o neoplasias benignas. La coexistencia de mutaciones dentro del CPT entre RET/PTC, RAS y BRAF es excepcional, aunque, como hemos visto anteriormente, en el 70% de los casos se encuentra por lo menos una de ellas8, 9. Por otra parte, se ha visto que hay asociación recíproca entre la edad y las mutaciones de BRAF y los reordenamientos del RET/PTC en el CPT, de forma que las primeras serían más frecuentes en individuos de mayor edad y éstas son más prevalentes en la edad pediátrica. Como se ha mencionado con anterioridad, la radiación ionizante es uno de los factores causales de RET/PTC y éste podría ser el motivo de que la población infantil sea mucho más proclive a presentar RET/PTC, como se ha visto en los casos aparecidos tras Chernobyl111, o en niños tratados con radioterapia externa112. Por el contrario, en la población adulta las mutaciones de BRAF son mucho más frecuentes, y no se ha hallado ninguna relación con las radiaciones107, 108, 113. Sin embargo, no se ha aclarado todavía por qué las mutaciones de BRAF ocurren en edades más adultas. Existen otras mutaciones de BRAF involucradas en la carcinogénesis, como la K601E o las que afectan al exón 11, pero que raramente se dan en el CPT109. De manera excepcional, en algunos casos asociados a la exposición a radiaciones ionizantes, se ha observado una fusión del gen BRAF con el gen AKAP9, por una inversión paracentromérica del brazo largo del cromosoma 7, la cual resulta en un oncogén, ACAP9-BRAF, que también induce una activación constitutiva de la vía de la MAPK110, 114, 115.
En los últimos 5 años se ha empezado a analizar la significación clínica de BRAF. El hecho de que se hayan observado mutaciones en las formas microscópicas de CPT107 indica, como en el caso de los receptores TK y de Ras, que la molécula se encuentra involucrada en las primeras fases de la carcinogénesis del CPT. De igual modo, la mutación se asocia a algunas variantes histológicas más agresivas del CPT, como la de células altas, y en cambio se encuentra con menor frecuencia en la variante folicular107, 109. Asimismo, la mutación de BRAF es más frecuente en las formas del CPT que sufren una desdiferenciación y evolucionan a carcinomas poco diferenciados y anaplásicos, contrariamente a lo que sucede en las formas poco diferenciadas, que evolucionan a partir de carcinomas foliculares106, 107. Por otro lado, varios estudios han investigado la utilidad de determinar la mutación de BRAF como factor pronóstico106,107,116-118. En la serie de Nikiforova et al107 se describe una correlación entre la mutación y la extensión extratiroidea en el momento del diagnóstico. En la serie japonesa de 126 casos, la mutación se asocia a estados más avanzados y a metástasis a distancia106. Sin embargo, no se ha visto ningún tipo de relación con los factores clínico-patológicos en dos series italianas y una americana108, 116, 117, aunque el número de casos analizados es menor. Xing et al109, 118, en una serie amplia, demuestran una asociación significativa entre la mutación de BRAF y la extensión extratiroidea, afección de ganglios regionales, metástasis a distancia, recurrencia tumoral y una pérdida de la diferenciación del tumor caracterizada por una disminución en la capacidad de captar 131INa. Los mismos autores apuntan la falta de estratificación de las diferentes variantes histológicas del CPT en los diversos estudios que, al incluirlas conjuntamente, explicarían la disparidad de estos resultados.
Finalmente, la exclusividad de la mutación de BRAF T1799A en el CPT ha hecho que se haya estudiado su utilidad en el diagnóstico de la enfermedad a partir de muestras de citología obtenidas por punción con aguja fina119-122. En todos los estudios, la especificidad y la sensibilidad para las muestras positivas para la mutación de BRAF han sido del 100%. Como la prevalencia de dicha mutación en estas series era del 44%, se puede afirmar que casi la mitad de los casos de CPT podría diagnosticarse preoperatoriamente mediante su análisis.
Aplicación terapéuticaAlrededor del 5% de los CPT experimentan un proceso de desdiferenciación que da lugar a neoplasias de agresividad elevada y muy mal pronóstico. En estos casos, el tratamiento convencional se ha demostrado inefectivo y otras estrategias, como la quimioterapia y la radioterapia, tampoco ofrecen resultados satisfactorios. En los últimos años, el descubrimiento de las bases moleculares causantes del CPT ha generado la posibilidad de actuar sobre nuevas dianas terapéuticas. La vía de señalización MAPK constituye uno de los objetivos más esperanzadores123. La mayoría de estas sustancias se encuentra todavía en fase experimental, pero muy probablemente pasarán a formar parte de los protocolos terapéuticos en un futuro no demasiado lejano83.
Inhibidores de las tirosincinasasExisten diferentes mecanismos para bloquear farmacológicamente el efecto de las TK. Entre ellos, el uso de moléculas de bajo peso molecular que actúan principalmente compitiendo con el lugar de unión de ATP124. Con el bloqueo de la actividad de fosforilación de las TK se consigue la inhibición de sus efectos enzimáticos en las vías de señalización intracelulares125. Como hemos visto, la destacada participación de distintos receptores con actividad TK, junto con el hecho de que su alteración sea un evento inicial en la oncogénesis del tumor, hace muy atractivo el uso este grupo de fármacos en el CPT, especialmente en casos resistentes al tratamiento habitual6. La mayoría de estos fármacos no muestran especificidad de substrato, sino que son potencialmente eficaces para inhibir diferentes RTK, especialmente los relacionados con la angiogénesis tumoral126. El dominio cinasa de RET/PTC presenta una homología del 50% con Kit, PDGFR (platelet-derived growth factor) y VEGFR (vascular endothelial growth receptor). Las moléculas que han mostrado una mayor especificidad para bloquear RET son las que, a priori, han despertado mayores expectativas127.
Vandetanib. Esta anilinoquinazolona es un potente inhibidor de VEGF, la TK considerada como principal regulador de la angiogénesis en el cáncer, y de EGFR, frecuentemente mutado en diferentes tipos de neoplasias128. A su vez, Carlomagno et al129 han demostrado que también es capaz de bloquear la actividad enzimática de RET, por lo que podría ser útil en el tratamiento del CPT.
Sorafenib. Recientemente, Henderson et al130 han demostrado en un modelo experimental de CPT que sorafenib es más eficaz en la reducción del crecimiento tumoral en casos portadores de RET/PTC que en los portadores de mutaciones de BRAF (a pesar de que la molécula fue diseñada originalmente como inhibidor de BRAF). En la actualidad hay en marcha diferentes estudios clínicos en fase II para valorar el efecto del fármaco en el cáncer de tiroides avanzado. Así, por ejemplo, Gupta-Abramson et al131 han tratado a 30 pacientes con cáncer de tiroides metastático y resistente al tratamiento con 131INa (de los que 18 eran CPT). Tras una media de 27 semanas de seguimiento, han observado una respuesta parcial en el 23,3% y una estabilización de la enfermedad en el 53,3% de los pacientes.
Sunitinib. Se ha demostrado su elevada eficacia para inhibir VEGF, PDGF y KIT. Además, Kim et al132 han publicado que es un potente inhibidor de RET/PTC en cultivos celulares de CPT. Desde un punto de vista clínico, Dawson et al133 han comunicado una respuesta favorable a la molécula tras 4 años de seguimiento en 2 pacientes con cáncer avanzado.
Los inhibidores de TK son una opción de futuro en el tratamiento del CPT avanzado o resistente a la terapia habitual. Para que ello sea posible es necesario definir su efectividad y diseñar moléculas que actúen de manera más específica para las distintas TK implicadas. En este sentido, empezamos a tener candidatos con mayor especificidad para RET/PTC134 o c-met135.
Bloqueadores de BRAF y MEKLos inhibidores de RET pueden ser efectivos en tumores portadores de RET/PTC, pero no cuando la mutación genética se encuentra por debajo de la cascada de señalización de la que constituye un elemento inicial. Mitsutake et al136 han demostrado experimentalmente que BRAF es necesario para la activación de MAPK inducida por RET/PTC. Así, el bloqueo de BRAF, por su posición en la vía, resulta más efectivo, sea cual sea el gen mutado10. La primera generación de moléculas inhibidoras de BRAF, como sorafenib, se caracterizaba por su bajo nivel tanto de especificidad como de potencia. El paso siguiente de los investigadores fue encontrar compuestos más específicos. Ello se consiguió con los bloqueadores de MEK, el elemento activado por BRAF. Actuar sobre moléculas más distales en la vía MAPK ofrece mayor selectividad de acción, lo cual reduce la toxicidad del fármaco. El primer inhibidor de MEK fue CI-1040118. Estudios posteriores demostraron que la molécula ofrecía una potencia insuficiente como antineoplásico en los diferentes tipos de tumores humanos en los que se testó. Posteriormente, Ouyang et al137 publicaron el efecto de dos nuevos inhibidores, AAL-881 y LBT-613, en líneas celulares de cáncer de tiroides portadoras de RET/PTC o con BRAF mutado. Ambas moléculas eran capaces de inhibir la fosforilación de MEK y ERK dependiente de Ras, BRAF y RET/PTC, reduciendo el crecimiento celular e induciendo apoptosis en las células malignas. A pesar de que los dos compuestos presentan una toxicidad que desaconseja su uso clínico, estos estudios demuestran que BRAF puede ser el talón de Aquiles del cáncer de tiroides138. Así, Mitsiades et al139 han demostrado que tanto el uso de AAL881 como la depleción de BRAF mediante la inhibición de su ARN resultan efectivos para frenar el crecimiento celular en cultivos de diferentes líneas de cáncer de tiroides. Además, el efecto es superior en las líneas portadoras de la mutación V600E que en aquellas con BRAF no mutado. A diferencia de los inhibidores de multiples cinasas iniciales, este nuevo grupo de compuestos bloqueadores de MEK no compiten en la región de unión a ATP, con 10 que aumenta su especificidad. Recientemente, se empezó a analizar una nueva generación de inhibidores de MEK. Entre las distintas moléculas sintetizadas, cabe destacar AZD6244. Su efectividad está siendo comprobada en líneas celulares de cáncer de tiroides140, pero están en marcha distintos ensayos clínicos en fase II en pacientes portadores de neoplasias de otras localizaciones, con resultados esperanzadores y un bajo potencial tóxico141.
En resumen, la vía de señalización de las MAPK constituye un elemento clave en la transformación neoplásica de las células foliculares tiroideas en el CPT. Específicamente, la alteración genética en tres componentes de la vía, los RTK, especialmente RET, Ras y BRAF se hallan en más del 70% de estos tumores. Dichas anomalías se consideran un paso inicial en la carcinogénesis y confieren algunas peculiaridades clínicas que pueden resultar útiles, sobre todo en el momento de clasificar a los pacientes según el pronóstico de la enfermedad a medio y largo plazo. Además, el conocimiento de las bases que rigen la oncogénesis del CPT nos da la posibilidad de actuar en ellas y aporta nuevas dianas terapéuticas en casos en que el tratamiento convencional resulta inefectivo.
