


Diabetes is a metabolic disease affecting approximately 300 million people worldwide. Neuropathy is one of its frequent complications, and may affect sensory, motor, and autonomic nerves. Its pathophysiology has not been fully elucidated. Several hypotheses have been proposed, and mitochondria have been suggested to play a significant role. This article reviews the mechanisms involved in mitochondrial dysfunction and development of diabetic neuropathy, consisting mainly of oxidative and inflammatory stress, changes in intracellular calcium regulation, apoptotic processes, and changes in mitochondrial structure and function that may lead to development of diabetic neuropathy.
La diabetes mellitus es una enfermedad metabólica que afecta a algo más de 300 millones de individuos en el mundo. Entre sus complicaciones, la neuropatía es frecuente y puede afectar nervios sensitivos, motores y autonómicos. Su fisiopatología no está totalmente esclarecida, se han propuesto varias hipótesis y el papel de la mitocondria ocupa un lugar importante. En este artículo, se revisan los mecanismos implicados en la disfunción mitocondrial y el desarrollo de la neuropatía diabética, que involucran principalmente el estrés oxidativo e inflamatorio, las alteraciones de la regulación del calcio intracelular, los procesos de apoptosis, y los cambios en la estructura y función de dicha organela que pueden llevar a la producción de neuropatía diabética.
Diabetes mellitus is a disease characterized by high blood glucose levels with insulin deficiency or resistance. Chronic hyperglycemia mainly affects the retina, kidneys, peripheral nerves, and cardiovascular system. Both the proximal and distal portions of sensory, motor, and autonomic nerves are affected by neuropathy. This may result in neuropathic pain, characterized by paresthetic and electric shock sensations, tingling, burning pain, and predominately distal and symmetrical lancinating pain, associated with thermal and mechanical allodynia. Gastrointestinal tract changes, including gastroparesis and intestinal pseudo-obstruction, erectile dysfunction, and predominately atrial cardiac arrhythmia may also occur. Foot ulcers of neuropathic origin leading to amputation may also result.1
The estimated worldwide prevalence of diabetes is 300million subjects.2 Today, 6.2% of the population in the United States have diabetes,3 and approximately 50% of these have some type of neuropathy. The risk of developing symptomatic neuropathy of the group of diabetic patients is approximately 4–10% at 5 years of diagnosis, and greater than 15% at 20years.4
The mechanisms leading to diabetic neuropathy have not been fully elucidated yet. It has been suggested that they result from hyperglycemia or the loss of insulin-dependent regulation. Different pathophysiological mechanisms have been postulated. One of them implicates the polyol pathway, where increased glucose leads to an increased activity of the enzyme aldose reductase and, thus, to the production of polyols which cause a decreased activity of the sodium/potassium (Na/K) pump and a depletion of reducing equivalents such as NADPH (nicotinamide adenine dinucleotide phosphate, reduced form) with a secondary decrease in glutathione production and, as a result, the induction of intracellular oxidative stress.5 Another potential mechanism implicates the glycosylation of proteins that cause an abnormal function in the nerve and the activation of receptors of products from advanced glycosylation (AGE), which are related to the expression of inflammatory mediators.6 A reduction in neurotrophic factors and circulating hormones, such as nerve growth factor (NGF) and neurotrophin-3 (NT-3), responsible for maintaining the normal phenotype of sensory neurons, has also been implicated.7 It has also been suggested that mitochondrial dysfunction may be a central mediator in diabetic neuropathy. Mitochondrion, the organelle responsible for oxidative phosphorylation, intended to produce adenosine triphosphate (ATP), is involved in different pathways for the generation of peripheral neuropathy, mainly in relation to its role in longer, sensory axons.8 The purpose of this article is to review these mechanisms, and to discuss and understand how they influence the development of neuropathy, how they interact, and their potential therapeutic implications.
Mitochondrial structure and functionMitochondrion is a cell organelle which apparently originated from a symbiotic relation between a bacterium and an eukaryotic cell approximately one thousand million years ago.9 Mitochondria measure 0.5–10μm, and their location in the different cell groups varies depending on energy requirements. They are very important in muscle, brain, and nerve.10 The organelle consists of an outer mitochondrial membrane (permeable to small molecules), an intermembrane space, an inner mitochondrial membrane (which is permeable to specific ions only), the cristae, and the mitochondrial matrix, where ions, metabolites, and mitochondrial deoxyribonucleic acid (DNA) are located.
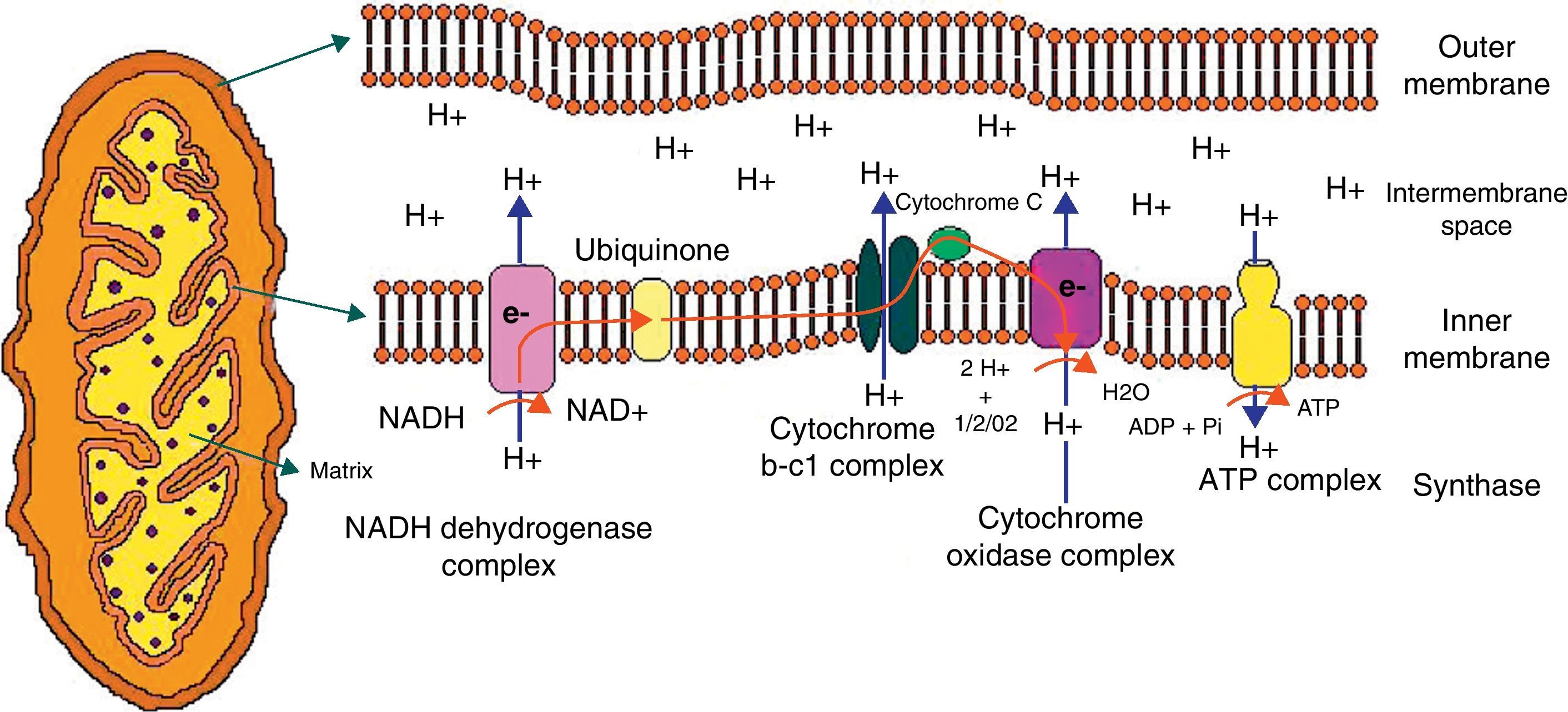
The mitochondrion is highly efficient in the utilization of oxygen and substrates mainly derived from glucose to produce cell energy in the form of ATP. Thus, electrons from oxidized substrates are transferred to oxygen by a series of reduction reactions to generate water.11 In this process, protons are pumped from the mitochondrial matrix to cross the inner mitochondrial membrane through the respiratory complexes forming the oxidative phosphorylation chain (Fig. 1).

Graphic representation of the mitochondrion and proton transfer through the mitochondrial membrane by the components of the oxidative phosphorylation chain.
Abbreviations used in the figure:
ATP, adenosine triphosphate; ADP, adenosine diphosphate; e, electrons; H, hydrogen; H2O, water; NAD, nicotinamide adenine dinucleotide; NADH, nicotinamide adenine dinucleotide reduced; Pi, phosphate; O2, oxygen.
The respiratory chain consists of five multipolypeptide enzyme complexes that form the oxidative phosphorylation system: complex I (NADH-ubiquinone reductase); complex II (FADH2 succinate-ubiquinone reductase); complex III (ubiquinol-cytochrome C reductase); complex IV (cytochrome C oxidase and two mobile electron transporters, ubiquinone and cytochrome C); and complex V (ATP synthase). Electrons generated from the reducing equivalents (NADH and FADH2) pass between the complexes and generate an increase in energy that allows for proton pumping to complexes I, III, and IV. Finally, the proton pump generated in the inner mitochondrial membrane is used to generate ATP.12
Although the vast majority of mitochondrial proteins (approximately 900) are produced by the nuclear genome and are imported to the mitochondrion, this organelle has a genome that is essential for respiratory function. The mitochondrial DNA has a size of 16kb, is circular, and contains 37 genes. Thirteen of these genes encode for protein subunits of the respiratory complexes I, III, IV, and V. Only complex II consists of proteins encoded for by nuclear genes.13
Mitochondrial functions vary along a spectrum that relates cell life and death. Functions include the production of reactive oxygen species (ROS), the opening of the permeability transition pore (PTP), cell respiration, ATP synthesis, and the production of proteins involved in apoptosis, such as the apoptosis-inducing factor (AIF).14 Mitochondrion also sequesters cytoplasmic calcium and regulates implications of this cation in cell function, thus maintaining calcium homeostasis by regulating direct calcium sequestration.
The mitochondrion carries out fusion and fission processes. The first such process consists of the binding of the outer and inner membranes to the mitochondrial matrix to produce a larger mitochondrion. Proteins called mitofusins, which are GTPases located in the outer membrane, participate in this process. These proteins are encoded by Mfn1 and Mfn2.15 The process opposite to fusion, mitochondrial fission, depends on the dynamin-related protein (Drp1), and creates multiple small mitochondria. The balance between mitochondrial fusion and fission is regulated in response to different stimuli. Thus, an increase in intracellular calcium increases fission, while a decreased mitochondrial movement results in a secondary decrease in mitochondrial fusion.16
Synaptic regions of axons have abundant mitochondria, which reflects the intensity of ATP demand to perform processes such as the fusion and recycling of ATP-dependent vesicles, as well as ATPase pumps that control the ionic environment in the synaptic membrane. Mitochondria are therefore mainly located in areas with a high energy demand such as pre- and post-synaptic domains, the initial axon segment to generate action potential, the nodes of Ranvier, growth cones, and free nerve endings. The locations described for mitochondria are often remote from the biogenetic site in neuron soma.17 Therefore, mitochondrial transport mechanisms exist to supply ATP and calcium buffer in the required areas. On the other hand, retrograde transport of damaged mitochondria to the cell body is required for their repair or degradation.
Mitochondria have processes for two-directional transport, stop, start, and change in direction. Long distance transport depends on ATP by way of motor proteins that displace along microtubules, while the actin cytoskeleton and neurofilaments are more important for anchoring and short distance movements of mitochondria.18 Forward transport is mediated by members of the kinesin superfamily (KIF),19 while retrograde transport of mitochondria is mediated by the protein dynein and accessory light chain proteins, including dinactin.
Mitochondria and diabetic neuropathyAs stated above, the prevalence rate of neuropathy in diabetic patients may be up to 50%.4,20 Several changes have been reported in peripheral nerves, including endoneural microangiopathy, axonal degeneration, loss of Schwann cells, paranodal demyelination, and loss of myelinated and unmyelinated fibers.21–24
The mitochondrion has been related to the pathophysiology of diabetic neuropathy.8 Studies in diabetic rats found structural changes in mitochondria, altered numbers of neurofilaments, and edema in axon terminals of nerves from the peripheral and autonomic nervous systems.25 Changes in mitochondrial function related to oxidative stress, dysfunction in intracellular calcium homeostasis, mechanisms involved in apoptosis, and axon transport abnormalities have also been reported.
Mitochondrial structure and diabetes mellitusThere are studies showing changes in Schwann cell mitochondria.26,27 One of them reported enlarged mitochondria, with a disruption or total absence of cristae, in the sciatic and sural nerves from rats fed diets very rich in galactose.27 Glycogen accumulation in the outer mitochondrial membrane was also shown in peripheral nerves.
Ultrastructural abnormalities in mitochondria are an important part in the sympathetic autonomic neuropathology in humans and a variety of mouse models.28 Such abnormalities include increased numbers of mitochondria and autophagic vacuoles, mixed with synaptic vesicles in sympathetic ganglia in both type 1 and type 2 diabetes.29,30
Mitochondria, oxidative stress, and diabetes mellitusReactive oxygen species (ROS) are generated by mitochondria as normal products of oxidation processes and are effectively removed by intrinsic antioxidant systems. When ROS accumulate, they are related to increased oxidative stress and impaired mitochondrial function. They have also been implicated as intermediates of apoptotic processes in mitochondrial DNA damage and neurodegeneration. This pathway is currently postulated to be a pathogenetic mechanism of sensory diabetic neuropathy.31,32
All components of the peripheral nervous system (sensory, motor, and autonomic nerves) are affected in diabetes, but neurodegeneration is most prominent in the longer axons of sensory neurons, where oxidative stress is considered to be a key pathological process that causes damage to the nerve.33,34
In cultured aortic endothelial cells, high intracellular glucose concentrations have been shown to increase electron delivery to the mitochondrial transport chain, causing mitochondrial hyperpolarization that leads to the production of reactive oxygen species.8 This mitochondrion-dependent process is proposed as a mediator of the complications of diabetes due to oxidative stress. The mechanism that explains the process is that high glucose concentrations increase NADH in mitochondria, and that this increase in electron availability or saturation causes oxygen reduction to superoxide radicals in the proximal part of the chain of electrons, which are associated with degenerative changes in mitochondrial structure.35 However, other studies in diabetic rats have shown that in neurons from the dorsal root ganglion (DRG) and in their axons, the inner mitochondrial membrane depolarizes and does not hyperpolarize.3 It has therefore been thought that in diabetes, a change in potential occurs in the inner mitochondrial membrane in populations of sensory neurons, resulting in changes in metabolic pathways and in the electron transport chain, probably caused by neurotrophic factor deficiency.
Treatment with insulin or NT-3 increases the delivery of reducing equivalents to the electron transport chain, with an improvement in membrane potential reduction by making it less negative and an additional increase in ATP synthesis.36 The mechanisms by which insulin and NT-3 cause stability in mitochondrial membrane potential include the phosphoinositide-3-kinase pathway (PI3-kinase). This pathway leads to the activation of factors such as protein kinase B and CREB (cAMP response element binding) transcription factor that regulate gene expression of factors in metabolic pathways and in the electron chain associated with neuronal mitochondrion.
In animals and humans, vascular dysfunction plays an essential role in the pathogenesis of diabetic neuropathy.32 This factor related to microangiopathy in the nerve includes impaired blood flow and endoneural hypoxia. Similarly, structural changes in nerve microvasculature help reduce endoneural perfusion, including the thickening of the basal membrane, pericyte degeneration, endothelial cell hyperplasia, the presence of abnormal arteriovenous communications, and vascular endothelial vulmerability.32 It is accepted that oxidative stress causes damage in diabetic neuropathy and is associated with a central role in nerve microangiopathy. These findings probably show the existence of a common point where mitochondrial dysfunction and subsequent oxidative stress may lead to and enhance vascular changes to generate the vascular damage responsible for diabetic microangiopathy: retinopathy, nephropathy, and neuropathy.35
Oxidative stress has been shown in sensory neurons and peripheral nerves by increased ROS production,35,37–39 lipid peroxidation,38 and protein nitrosylation.40,41 These result in energy dysfunction in the nerve with a reduction in highly energetic intermediates, impaired axonal transport of proteins, and poor function of ion pumps.42 The ROS produced may damage mitochondrion and its DNA. The accumulation of this damage in the nerve reduces nerve function, and these changes in mitochondrial dynamics are related to a decreased axon count and accumulation in the somas of DRG neurons.
In conclusion, oxidative stress is associated with increased ROS production, which secondarily leads to mitochondrial dysfunction, processes which are probably implicated in the development of diabetic neuropathy. Secondary nerve changes include the reduction of energy intermediates, lipid peroxidation processes, and decreased mitochondrial count, amongst others. Hyperglycemia and oxidative stress have also been related to changes in the value of mitochondrial membrane potential in dorsal root ganglion neurons, which improves after the administration of insulin and neurotrophic factors. On the other hand, there is a common pathway that combines oxidative stress with microangiopathy to increase nerve damage. Thus, no single causative process exists, but the addition and enhancement of these processes accounts for the pathogenesis.
Mitochondrion, calcium, and diabetes mellitusAs previously noted, the mitochondrion plays various roles to maintain intracellular homeostasis, including calcium regulation. There are sensors for calcium transport as a physiological response. Free calcium levels in the cell range from 50 to 100nM in cytosol and approximately 0.5–1mM in endoplasmic reticulum lumen, so that any change leading to the elevation of these levels in cytosol may have pathological consequences, including cell death.42,43 For this reason, mitochondrial dysfunction is associated with changes in calcium homeostasis, and a major role of these changes in the pathophysiology of diabetic neuropathy has been established.44,45
In experimental studies of diabetes induced by streptozotocin (STZ) and diabetic patients, abnormalities in intracellular calcium balance have been seen in dorsal root ganglion neurons, as well as in smooth muscle, secreting cells, and mouse osteoblasts.3 These changes consist of an increased intracellular calcium concentration, a decreased activity of calcium transporters, and a decrease in calcium-evoked signals.
In neurons from rats with STZ-induced diabetes, intramitochondrial calcium concentration may increase due to changes in transporters. This change in calcium homeostasis may stimulate a greater production of ROS inside the mitochondrion and, secondarily, the generation of oxidative stress. The cause of this phenomenon is related to an increase in the tricarboxylic acid cycle and oxidative phosphorylation caused by increased intracellular calcium levels, which makes mitochondrial work more rapid and consumes more oxygen, with a concomitant increase in ROS.46–49
In vivo and in vitro studies have shown that the critical factor in mitochondrial dysfunction is not hyperglycemia, but a deficiency of insulin and neurotrophic factors, which are responsible for maintaining mitochondrial membrane potential and for increasing ATP synthesis.50 In the above circumstances, treatment with insulin and neurotrophic factors normalized mitochondrial membrane polarity and intracellular calcium levels.36
Increased intracellular calcium levels may cause complete or partial depolarization of the inner mitochondrial membrane. This depolarization is prolonged in DRG neurons from rats with diabetes and may be blocked with carbonylcyanide-m-chlorophenylhydrazone (CCCP), an agent that promotes calcium binding to the mitochondrion. This shows the significance of the buffer function performed by the mitochondrion with calcium in diabetic neurons.51
On the other hand, decreased ATP levels in dysfunctional mitochondria may lead to a lower activity of Na/K ATPase pumps, which results in an increase in intracellular calcium and an inversion of the Na/Ca exchanger, with a secondary increase in intracellular calcium which is not sequestered by the mitochondrion and is therefore deleterious, finally leading to axon degeneration.
In conclusion, impaired calcium homeostasis due to mitochondrial dysfunction is a factor related to diabetic neuropathy, with findings showing increased intracellular concentrations and an imbalance in calcium transport in DRG neurons from rats and diabetic patients. This condition is associated with mitochondrial depolarization, ion pump dysfunction, and the generation of ROS. Some of these conditions related to calcium regulation may improve with insulin therapy.
Mitochondrion, apoptosis, and diabetes mellitusThe mitochondrion has a predominant role in apoptosis, one of the pathways of programmed cell death. This organelle is involved in this process by promoting the translocation of cytochrome C (proapoptotic) to cytosol, and may secondarily induce apoptosis. On the other hand, antiapoptotic proteins are also found in the mitochondrion, so that a crosstalk exists between life and death mediated by it. The activation of apoptosis is associated with sensory diabetic neuropathy by a concomitant mitochondrial dysfunction.52
Studies in the cultures of embryonal rat sensory neurons show that increased glucose concentrations (45nM) cause chronic mitochondrial depolarization with ATP depletion, followed by apoptosis resulting from mitochondrial dysfunction and secondary activation of caspases 3 and 9. In this same study, glucose increase in neurons was associated with greater local production of ROS as a finding prior to the induction of apoptosis.53
Loss of unmyelinated neurons occurs in models of rats with type 1 diabetes, but no information supporting the presence of apoptosis or abnormal mitochondria is available.54 However, other studies in neurons from diabetic rats showed impairment in axon morphology and growth caused by elevated glucose levels, which associated with oxidative stress processes resulted in axon degeneration but not in cell death.55 These results suggest the potential existence of different response mechanisms to hyperglycemia in axons as compared to neuronal bodies and similarly to the mitochondria found at each of these sites. This may explain differences in the results of these studies.
The loss of factors such as AKT (also called PKB/Akt, serine/threonine kinase) in diabetes induces the translocation of proapoptotic factors to the outer mitochondrial membrane and is important in neurodegeneration in embryonal and mature neurons in which mitochondrial depolarization is involved as a process preceding apoptosis.56
Studies in rats with STZ-induced diabetes showed decreases in peripheral nerve conduction velocities as compared to control rats, as well as greater apoptosis in DRG neurons from diabetic rats. Mitochondrial membrane potential was also more positive in these rats as compared to controls, and the restoration of euglycemia in 2 weeks decreased apoptosis and normalized mitochondrial membrane potential.57 In this same study, levels of antiapoptotic Bcl2 proteins (isolated from B2 lymphoma cells) decreased in sensory neurons from diabetic rats, and translocation of cytochrome C from the membrane to cytoplasm with induction of apoptosis occurred.
In conclusion, some studies allow us to relate apoptosis and mitochondrial dysfunction to the development and pathophysiology of diabetic sensory neuropathy. However, other studies do not conclusively show this relationship. Discrepancies in the different results may be attributed to the different animal models used, and also to the methodology and follow-up time used. The proposed mechanisms involved include the chronic depolarization of the mitochondrial membrane which precedes apoptosis secondary to glucose increase in DRG. In addition, decreased antiapoptotic protein levels have been found, and translocation of proapoptotic factors to the cytosol has been seen.
Axonal transport, mitochondrion, and diabetes mellitusMitochondria are located in the sites with the greatest energy requirements. The transport of mitochondria mediated by proteins and dependent on the abovementioned factors, including ATP levels, calcium, and fusion/fission processes, is therefore required. Although the normal location of mitochondria in the axon has been reported, different mitochondrial locations have been documented in pathological conditions such as some neuropathies, where they accumulate in unmyelinated axon segments and regions of disruption of the axoglial junction.58
Decreased ATP production secondary to mitochondrial dysfunction in diabetes may result from hypoinsulinemia23 and leads to decreased axonal transport of proteins and reduced protein synthesis. These combined factors are associated with distal axon degeneration.
Impaired axonal transport of mitochondria leads to mitochondria accumulation. These mitochondrial aggregates may contribute to the development of local pathology as the result of increased ROS production, loss of normal energy production, and abnormal calcium regulation.59
The impact of high glucose concentrations on mitochondrial traffic and associated lesions has not been elucidated. A study of rat liver cells and myoblasts showed that mitochondrial fragmentation is a process induced by glucose required for mitochondrial respiration and ROS production.60 On the other hand, the treatment of pancreatic beta cells and neurons with glucose results in mitochondrial fragmentation/fission and, in some scenarios, this process is followed by cell death and apoptosis.61
To sum up, no direct results showing a clear relationship between diabetic neuropathy and mitochondrial transport are available. However, mitochondrial dysfunction has indirectly been related to decreases in ATP levels and axonal transport of proteins, and with the accumulation of mitochondria in neuron bodies, which suggests changes in transport along the axon.
ConclusionDiabetic neuropathy is a complication with a significant impact where sensory, motor, and autonomic nerves are compromised. The polyol pathway and protein glycosylation have traditionally been proposed as its causative mechanisms. Mitochondrial dysfunction has been considered as another causative agent of diabetic neuropathy.
Mitochondrial changes include increased generation of ROS, decreased mitochondrial membrane potential, dysfunction in intramitochondrial calcium regulation, the depletion of ATP production, and resultant defects in axon transport and an increase in products that may lead to apoptosis. These mechanisms, combined, may lead to axon degeneration. These findings predominate in longer nerves. The reasons for the predominance of mitochondrial compromise in dorsal root ganglia and sensory nerves have not been elucidated yet.
The pathophysiology explaining the causative mechanisms of diabetic neuropathy has not been defined yet. Several studies have been conducted in animal models, mainly mice, and further research in humans is therefore required. However, there are limitations such as difficulties in taking biopsies, the risks inherent in the procedure, and lack of access to special technology, which make the conduct of studies more difficult.
The role played by mitochondrial changes in the pathophysiology of diabetic neuropathy should facilitate the future search for new therapeutic resources aimed at correcting mitochondrial dysfunction and improving the quality of life of diabetic patients.
Conflicts of interestThe authors state that they have no conflicts of interest.
Please cite this article as: Hernández-Beltrán N, et al. El papel de la mitocondria en el dolor de la neuropatía diabética. Endocrinol Nutr. 2013;60:25-32.
