




The use of protease inhibitors (PI) has led to a decrease in HIV-1-related mortality and morbidity. The objective of this study was to collect safety data on treatment with fosamprenavir/ritonavir (FPV/r) 700/100mg BID in HIV-infected patients through an expanded access program.
Patients and methodsProspective, multicenter, noncomparative study in HIV-1 infected adults, for whom a regimen containing FPV/r 700/100mg BID was appropriate.
ResultsA total of 678 patients were included in the intention-to-treat (ITT) and safety population. The on-treatment (OT) population contained 587 patients: 76% male, 98% Caucasian, and median age 41 years. Median CD4 cell count was 351cells/μL, HIV-RNA was 3logcopies/mL, and 49% of patients were in CDC class C. After 24 weeks of treatment, serum viral load decreased a median of 1.3logcopies/mL and 73% of patients had <400copies/mL (P<.0001 vs. baseline); 48-week results were similar. CD4 cell count increased a median of 49 and 62cells/μL at 24 and 48 weeks, respectively. Adverse events (AEs) associated with the study medication occurred in 21% of patients.
ConclusionsRitonavir-boosted fosamprenavir as part of antiretroviral therapy is a potent, safe treatment in real-life clinical circumstances.
los Inhibidores de proteasa (PI) tuvieron un impacto positivo en la disminución de la morbilidad y mortalidad relacionada con infección por el VIH-1. El objetivo de este estudio fue obtener información de seguridad sobre fosamprenavir/ritonavir (FPV/rtv) 700/100mgBID mediante un Programa de Acceso Expandido (EAP).
Pacientes y métodosestudio prospectivo, multicéntrico y no comparativo, en adultos infectados por VIH-1 en los que un régimen conteniendo FPV/rtv 700/100mg BID se considerase adecuado.
Resultadosun total de 678 sujetos fueron incluidos en la población por intención de tratar (ITT) y de seguridad. Por protocolo (OT) se incluyó a 587 sujetos, un 76% varones, un 98% caucásicos y con una mediana de edad de 41 años. La mediana de CD4 fue 351 células/μl, de VIH-ARN 3 log copias/ml y un 49% en clase C de los CDC. Tras 24 semanas de tratamiento, la carga viral disminuyó 1,3 log copias/ml (mediana) y un 73% tenía<400 copias/ml (p<0,0001 frente a basal), al igual que en semana 48. Los CD4 aumentaron 49 y 62 células/μl en semana 24 y 48, respectivamente. Acontecimientos adversos (AE) relacionados con la medicación del estudio aparecieron en un 21% de los sujetos.
Conclusionesfosamprenavir potenciado con ritonavir como parte del tratamiento antirretroviral resultó ser un potente y seguro tratamiento antirretroviral bajo las condiciones clínicas habituales.
Protease inhibitors (PI) have had a dramatic, positive impact on HIV-related mortality and morbidity in HIV-infected individuals.1,2 When a PI is considered for initial therapy, current guidelines recommend ritonavir-boosted PIs as the appropriate option.3,4 Fosamprenavir/ritonavir-based regimens have shown non-inferiority compared to nelfinavir or lopinavir/ritonavir in naïve populations.5,6 In pretreated patients, the use of fosamprenavir/ritonavir (FPV/r) versus lopinavir/ritonavir resulted in similar percentages of patients with HIV RNA levels <50copies/mL.7 Thus, it would be of value for physicians to have more information on treatment with fosamprenavir/ritonavir under real-life conditions.
The main objective of this study was to collect safety information on treatment with fosamprenavir 700mg/ritonavir 100mg (FPV/r 700/100mg) BID in HIV-1-infected patients who received this regimen through an expanded access program, according to approved conditions.
Patients and methodsPatientsThe fosamprenavir/ritonavir expanded access program was started in May 2004 and completed in October 2005. The inclusion criteria were as follows: HIV-1 infected adults (⩾18 years old), HIV-RNA ⩾1000copies/mL, CD4 cell count ⩽350cells/mL, and lack of an alternative HAART regimen or currently taking amprenavir or poor adherence to current treatment. Patients were excluded if they had intolerance or viral resistance to FPV/r, baseline laboratory abnormalities precluding patient's participation, known hypersensitivity to FPV or ritonavir (RTV), concomitant medication that contraindicated the use of FPV or RTV, or any serious hepatic disorder, or if they were pregnant or breast-feeding women. Participants provided written informed consent to participate, and women of childbearing potential were requested to have a negative pregnancy test and use a proven method of contraception.
Study designThis was a prospective, multicenter, non-comparative, open-label study, conducted in 86 centers in Spain. The study protocol was reviewed and approved by the corresponding research ethics committees and the Agencia Española de Medicamentos y Productos Sanitarios (Spanish Agency for Medicines and Medical Devices) and was carried out in accordance with International Conference on Harmonization (ICH) Good Clinical Practice guidelines, and the Declaration of Helsinki. Demographics and adverse events were recorded at baseline visit. Plasma HIV-RNA, CD4 cell count, hematology, and clinical chemistry analyses were performed at baseline (day 1) and every 3 to 5 months thereafter (according to daily clinical practice). Genotyping and phenotyping were performed in cases of antiretroviral treatment failure. All laboratory analyses for the study were carried out at local sites. Patients were withdrawn from the study if treatment-limiting adverse events occurred, treatment was self-interrupted, there was no evidence of clinical benefit, HIV disease progressed, or 30 days had passed since FPV came on the market in Spain. Each patient received 1 tablet of FPV 700mg plus 1 RTV 100mg soft gel capsule twice daily plus 2 or more antiretroviral agents. Efficacy was assessed by the change in log10 plasma HIV-RNA copies/mL at week 24 versus baseline, by the number and percentage of patients achieving HIV-RNA plasma levels <400copies/mL (“undetectable”) and by median CD4+ cell count change.
Adverse events were recorded and classified into 3 categories of severity (mild, moderate and severe). Abnormal laboratory findings were recorded and included serum levels of triglycerides >499mg/dL, total cholesterol >240mg/dL, and HDL cholesterol <40mg/dL.
Statistical analysesBecause of the characteristics and objective of the study, no formal sample size calculation was needed. Descriptive methods were used, with some exceptions when comparisons were feasible or considered clinically relevant. The ITT population included patients who received at least one dose of study medication and attended at least 2 visits. The ITT population excluding major protocol violators and those with no viral load measurement during follow-up constituted the OT population. SAS, v 9.1, was used for the analyses.
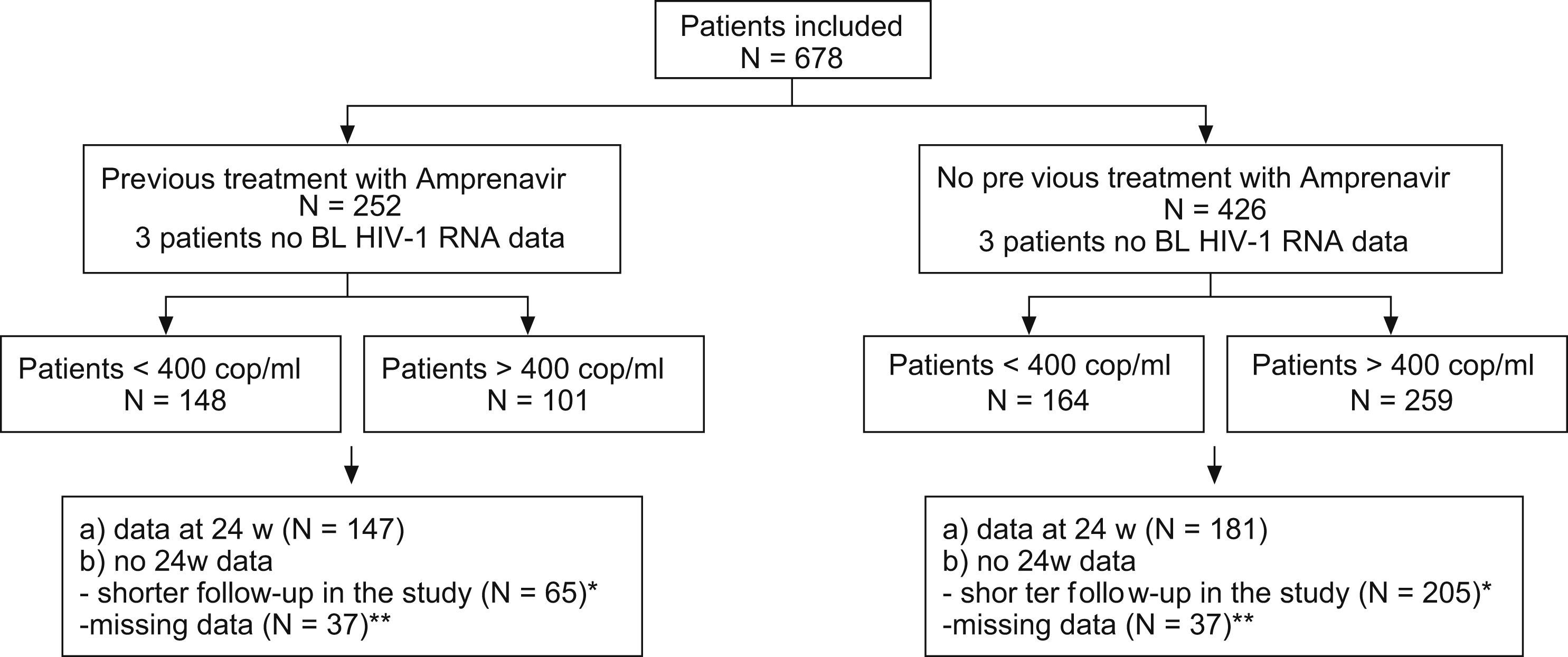
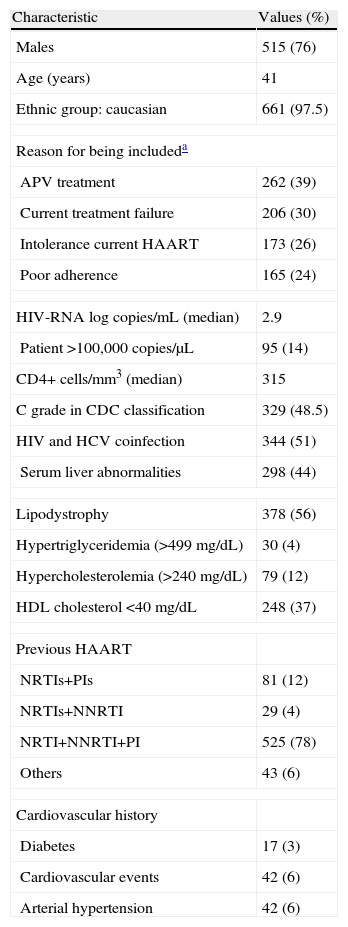
ResultsA total of 678 patients comprised the ITT efficacy and safety population. Excluding withdrawals and those with missing data, the OT population included 587 patients (74%). Distribution of patients according to amprenavir use, 24-week data, and viral load is shown in Fig. 1. Baseline characteristics are summarized in Table 1. The most frequent treatment backbones selected during the study was 2 nucleoside reverse transcriptase inhibitors (NRTIs) (33.8%) or an NRTI plus a non-nucleoside reverse transcriptase inhibitor (NNRTI) (45.6%).

Baseline characteristics of the patients
| Characteristic | Values (%) |
| Males | 515 (76) |
| Age (years) | 41 |
| Ethnic group: caucasian | 661 (97.5) |
| Reason for being includeda | |
| APV treatment | 262 (39) |
| Current treatment failure | 206 (30) |
| Intolerance current HAART | 173 (26) |
| Poor adherence | 165 (24) |
| HIV-RNA log copies/mL (median) | 2.9 |
| Patient >100,000 copies/μL | 95 (14) |
| CD4+ cells/mm3 (median) | 315 |
| C grade in CDC classification | 329 (48.5) |
| HIV and HCV coinfection | 344 (51) |
| Serum liver abnormalities | 298 (44) |
| Lipodystrophy | 378 (56) |
| Hypertriglyceridemia (>499mg/dL) | 30 (4) |
| Hypercholesterolemia (>240mg/dL) | 79 (12) |
| HDL cholesterol <40mg/dL | 248 (37) |
| Previous HAART | |
| NRTIs+PIs | 81 (12) |
| NRTIs+NNRTI | 29 (4) |
| NRTI+NNRTI+PI | 525 (78) |
| Others | 43 (6) |
| Cardiovascular history | |
| Diabetes | 17 (3) |
| Cardiovascular events | 42 (6) |
| Arterial hypertension | 42 (6) |
APV: amprenavir; HAART: highly active antiretroviral therapy; HIV: human immunodeficiency virus infection; HCV: hepatitis C virus infection; NRTI: nucleoside reverse transcriptase inhibitor; NNRTI: non-nucleoside reverse transcriptase inhibitor; PI: protease inhibitor; CDC: Centers for Disease Control and Prevention.
A total of 650 patients who had at least two serum viral load result in addition to the baseline value comprised the ITT population for efficacy. Mean (range) follow-up time was 24 weeks (2.4–71.2 weeks). After this time, viral load decreased a median of 1.3log HIV-RNA copies/mL. This reduction was higher (2.05logcopies/mL) in patients with serum HIV-RNA >400copies/mL, but was only statistically significant for those with HIV RNA⩾3logcells/mL at baseline (P<.0002). In centers with lower viral load detection thresholds (n=68), 77% of patients with >400 HIV-RNA copies/mL at baseline reached <50 HIV-RNA copies/mL after 24 weeks of treatment. At 48 weeks (n=150), 73% of patients presented <400copies/mL compared with 46% at baseline (P<.0001). At 48 weeks, 93% of patients virologically controlled at the time of study entry remained virologically suppressed. No PI mutations were detected in patients failing antiretroviral treatment; however, half of them showed lower viral load values compared to baseline.
Median CD4 cell count change was not statistically significant (increase of 49cells/μL at 24 weeks and 62 cells at 48 weeks). The changes were similar in patients with baseline CD4 count <200cells/μL (39cells/μL at 24 weeks and 87cells/μL at 48 weeks).
SafetyAmong the total, 51.2% of patients experienced adverse events. There were 685 adverse events in 347 patients and 9% of them were classified as grade III–IV. The most common adverse events were diarrhea (62 events 9.1%), hypertriglyceridemia (39 events 5.8%), flu syndrome (37 events 5.5%), hypercholesterolemia (23 events 3.4%), fever (21 events 3.1%), respiratory infections (21 events 3.1%), vomiting (18 events 2.7%), nausea (18 events 2.7%), and ALT/AST elevation (14 events 2.1%).
Adverse events related to the study medication occurred in 19% of patients, and mainly included diarrhea (30 events, 4.4%), hypertriglyceridemia (25 events, 3.6%), hypercholesterolemia (19 events, 2.8%), nausea (12 events, 1.8%), vomiting (7 events, 1.0%), and ALT/AST elevation (7 events, 1.0%). The study medication was interrupted because of adverse events in only a small number of patients (19; 2.8%).
Seventy-six patients (11%) developed 114 serious adverse events, but only 12 of these events (in 7 patients) were possibly related to the study drug. Five patients died of causes unrelated to the study medication.
The number of patients with hematological alterations at completion of the study was similar to the number at baseline. No changes were found for serum glucose, amylase or albumin levels. The percentage of patients with AST elevations decreased from 29.4% to 22.9% (P=.02). After 24 and 48 weeks of treatment, there was an increase in the number of patients with serum levels of cholesterol >240mg/dL (P<.001) or HDL-cholesterol >40mg/dL (P<.05), and no changes in hypertriglyceridemia (>499mg/dL).
DiscussionData on FPV/r use are available from clinical trials5–7; however, there is no published information to date on FPV/r use in routine clinical practice. As compared with previous FPV/r studies, the patients in this investigation had a lower baseline viral load (median viral load in the KLEAN study was 5.1log copies/mL) and half of them presented adequate virological control at the beginning of the study. In addition, there was a higher percentage of Caucasians, CDC class-C status, and HCV-HIV coinfection. However, patients were comparable in terms of sex and age,5–7 and 25% of patients had CD4 cell count <200cells/μL. Therefore, by selecting only patients that presented a detectable viral load at baseline, it is not surprising that the efficacy results were comparable or better than those of previous clinical trials (viral load became undetectable at week 24 in 77%).6–8 In line with previous data,9 no new PI resistance mutations were detected in failing patients, providing new opportunities for the use of other PI-containing regimens. Although the backbone medication can have an impact on efficacy results, the backbone used in this study did not differ that of from previous studies (2 NRTIs or 1 NRTI+1 NNRTI), and it is worth mentioning that most patients had been treated previously with PIs, as is common in heavily treated HIV populations. The present study supports previously reported safety results for FPV/r.5–7,10 There were fewer discontinuations due to adverse events than in other studies (2.8% vs. 5%);6 nevertheless, it should be taken into account that almost 40% of patients were tolerant to amprenavir. However, bearing in mind that overall tolerability in HIV/HCV coinfected patients is lower and that the percentage of coinfected patients in this study was higher (51%) compared with a range of 10%–15% in other studies,5–7 we can conclude that FPV/r was well tolerated in this study.
In our opinion, the fact that the inclusion criteria and visiting schedule were not restricted in this study, in line with normal clinical practice, indicates that FPV/r-containing regimens can be considered efficacious and safe in patients receiving this therapy in real-life conditions.
The following are the members of the Fosamprenavir Expanded Access ProgramAguirrebengoa, K. (Hospital de Cruces, Bilbao); Alemán, R. (Hospital Universitario de Canarias, Tenerife); Alonso, C. (Hospital Universitario Sant Joan de Reus, Tarragona); Orti, A. (Hospital Virgen de la Cinta, Tarragona); Arazo, P. (Hospital Miguel Servet, Zaragoza ); Arrizabalaga, J. (Hospital de Aranzazu, San Sebastián); Bachiller Luque, P. (Hospital Río Hortega, Valladolid); Berdun, M.A. (Hospital San Jorge, Huesca); Báguena, F. (Fundació Sanitaria d’Igualada, Barcelona); Blanco, F. (Hospital Carlos III, Madrid); Boix, V. (Hospital General de Alicante); Blanquez, R.M. (Hospital Morales Messeguer, Murcia); Cano, A. (Hospital General de Murcia); Carmena, J. (Hospital Dr. Peset, Valencia); Causse, M. (Hospital Carlos Haya, Málaga); Clotet, B. (Hospital Germans Trias i Pujol, Barcelona); Cucurull, J. (Hospital Figueres, Girona); Cuadrado, J.M. (Hospital San Juan de Alicante); Chocarro, A. (Hospital Virgen de la Concha, Zamora); Dalmau, D. (Hospital Mutua de Terrassa, Barcelona); Del Pozo, M.A. (Hospital Clínico de Valladolid); Domingo, P. (Hospital de Sant Pau, Barcelona); Echeverría, S. (Hospital Marqués de Valdecilla, Santander); Flores, J. (Hospital Arnau de Vilanova, Valencia); Force, L. (Hospital de Mataró, Barcelona); Francés, A. (Hospital Insular de las Palmas de Gran Canaria); Galindo, M.J. (Hospital Clinico Universitario, Valencia); Gálvez, J. (Hospital Virgen de Macarena, Madrid); Gálvez, M.C. (Hospital de Torrecárdenas, Almería); García, J.L. (Hospital Santa María la Rosell, Murcia); Guerrero, F. (Hospital Puerta del Mar, Cádiz); Górgolas, M. (Fundación Jiménez-Díaz, Madrid); Gutiérrez, F. (Hospital de Elche); Hayek, M. (Hospital Nuestra Señora de la Candelaria, Tenerife); Hernández, J.J. (Complejo Hospitalario de Jaén); Hernández, J. (Hospital San Cecilio, Granada); Jusdado, J.J. (Hospital Severo Ochoa, Madrid); Kindelán, J.M. (Hospital Reina Sofía, Córdoba); Knobel, H. (Hospital del Mar, Barcelona); Lorenzo, J.F. (Hospital General Yagüe, Burgos); Llibre, J.M. (Hospital Sant Jaume, Calella, Barcelona); López, J. (Hospital La Fe, Valencia); Lozano, F. (Hospital de Valme, Sevilla); Mallolas, J. (Hospital Clínic, Barcelona); Maradona, J.A. (Hospital Nuestra Señora de Covadonga, Oviedo); Mariño Callejo, A. (Hospital Arquitecto Marcide, La Coruña); Márquez, M. (Hospital Virgen de la Victoria, Málaga); Martín, C. (Hospital Nuestra Señora de la Montaña, Cáceres); Mascaró, J. (Hospital Dr. Josep Trueta, Girona); Miralles, P. (Hospital Gregorio Marañón, Madrid); Morano, L. (Hospital Meixoeiro, Pontevedra); Muñoz, A, (Hospital Infanta Cristina, Badajoz); Ocampo, A. (Hospital Xeral de Vigo, Pontevedra); Ojea, R, (Hospital de Pontevedra); Ortega, E. (Hospital General Universitario, Valencia); Oteo, J.A. (Hospital Provincial de la Rioja, La Rioja); Pasquau, F. (Hospital de la Marina Baixa, Alicante); Pasquau, J. (Hospital Virgen de las Nieves, Granada); Pedreira, J.D. (Hospital Juan Canalejo, La Coruña); Pedrol, E. (Hospital de Granollers, Barcelona); Peralta, G. (Hospital Sierrallana, Santander); Pérez, M.J. (Hospital Ramón y Cajal, Madrid); Peña, J.M. (Hospital La Paz, Madrid); Podzamcer, D. (Hospital Bellvitge, Barcelona); Portu Zapirain, J. (Hospital Txagorritxu, Vitoria); Prada, J.L. (Hospital Costa del Sol, Marbella); Prieto, A. (Hospital Santiago de Compostela, La Coruña); Pujol, E. (Hospital Juan Ramón Jiménez, Huelva); Pulido, F. (Hospital 12 de Octubre, Madrid); Redondo, C. (Hospital Universitario Virgen de la Arrixaca, Murcia); Ribera, E. (Hospital Vall d’Hebron, Barcelona); Riera, M. (Hospital Son Dureta, Palma de Mallorca); Rodríguez, R. (Hospital de Orense); Roca, V. (Hospital Clínico, Madrid); Rubio, R. (Hospital 12 de Octubre, Madrid); Rubio, M. (Hospital Arnau de Vilanova, Lleida); Sala, M. (Hospital Parc Tauli, Barcelona); Santamaría, J.M. (Hospital de Basurto, Bilbao); Sanz-Moreno, J. (Hospital Príncipe de Asturias, Alcalá de Henares, Madrid); Sanz, J. (Hospital La Princesa, Madrid); Silvariño, R. (Hospital de San Eloy, Vizcaya); Soriano, V. (Hospital Carlos III, Madrid); Telenti, M. (Hospital Central de Asturias, Oviedo); Terrón, J.A. (Hospital SS de Jerez); Viciana, P. (Hospital Virgen Rocío, Madrid); Vilades, C. (Hospital Joan XXIII, Tarragona); Zarzalejos, J.M. (Hospital Dr. Negrín, Tenerife).
On behalf of the Fosamprenavir Expanded Access Program Group, the authors wish to thank the patients enrolled in the study; without their contribution, this data would never have been collected. We also thank Diana Rodriguez for her administrative support.
Potential conflicts of interest
Isabel Luque and Felipe Rodriguez belong to the Medical Department of GlaxoSmithKline in Spain. All other authors declare no conflicts of interest.

