





La norma UNE-EN-ISO 15189:2007. Laboratorios clínicos. Requisitos particulares para la calidad y la competencia especifica los requisitos de gestión y los requisitos técnicos que deben cumplir los laboratorios de microbiología clínica que quieran alcanzar un máximo de calidad en la realización de análisis microbiológicos. Con la implantación de esta norma se puede conseguir la acreditación o el reconocimiento formal por una entidad autorizada de la aptitud del laboratorio para realizar un ensayo o un conjunto de ensayos. En España la entidad evaluadora es la Entidad Nacional de Acreditación.
El objetivo de esta revisión es acercar los requisitos de la norma UNE-EN-ISO 15189:2007 a los laboratorios de microbiología, con un enfoque práctico y orientado a los estudios de bacteriología y serología. Se definen brevemente los alcances y se especifican los requisitos que ha de cumplir el análisis microbiológico, el control de la documentación, el aseguramiento de la calidad, el control de los equipos, la gestión del personal, los sistemas de información, suministros y otros servicios externos y, por último, se indican los sistemas de evaluación para monitorizar la mejora continua de los procesos y del servicio prestado por el laboratorio.
The UNE-EN-ISO 15189:2007 standard specifies the management and technical requirements that clinical microbiology laboratories must meet to achieve optimal quality when performing microbiological analyses. With implementation of this standard, a laboratory can receive the accreditation and formal recognition of an authorized body, certifying that it is apt for performing an assay or group of assays. In Spain, laboratories that apply these standards can be accredited by the Entidad Nacional de Acreditación (ENAC, Spanish accreditation body).
The purpose of this review is to familiarize clinical microbiology laboratory specialists with the UNE-EN-ISO 15189:2007 standard through a practical approach focussed on bacteriology and serology studies. We briefly define the scope and specify the requisites required for managing the quality of the procedures and processes involved in performing tests on human specimens, for document control, and for management of instruments and equipment, personnel, information systems, supply systems, and external services. Lastly, evaluation approaches are indicated to achieve continuing improvement of the processes carried out and the services the laboratory provides.
Actualmente existe una apuesta clara por alcanzar y por demostrar la calidad en los servicios sanitarios, fomentándose desde la administración la evaluación periódica y externa de la calidad y la seguridad de los centros mediante auditorías por parte de instituciones públicas o privadas que garanticen una evaluación independiente1. En este marco, la acreditación es el reconocimiento formal por parte de un organismo autorizado de que un laboratorio clínico es competente para realizar un ensayo o un conjunto de ensayos2. En España el organismo evaluador autorizado es la Entidad Nacional de Acreditación.
Los laboratorios de microbiología que quieran acreditarse deben cumplir los requisitos establecidos en la norma UNE-EN-ISO 15189:2007. Laboratorios clínicos. Requisitos particulares para la calidad y la competencia. La implantación de un sistema de gestión de calidad (SGC) basado en esta norma garantiza que los ensayos se llevan a cabo con un alto grado de calidad y debe acompañarse de una mejora en el servicio ofrecido al usuario3. La norma incluye los requisitos de gestión exigidos por la norma ISO 9001:20004, y además incluye la evaluación de la competencia técnica del laboratorio, haciendo hincapié en aspectos como la cualificación y la competencia del personal, la adecuación de las instalaciones, el uso de métodos validados y equipos controlados, la gestión de la información y la participación en programas de intercomparación2.
En la norma UNE-EN-ISO 15189:2007 se describen conceptos como alcance, trazabilidad, incertidumbre, no conformidad, validación, verificación y otros, con los que el microbiólogo clínico no está familiarizado. El objetivo de esta breve revisión es acercar los requisitos exigidos por esta norma a los laboratorios de microbiología y hacer una serie de recomendaciones para facilitar su implantación, orientada a los estudios de bacteriología y serología, aunque también puede aplicarse a algunos ensayos de microbiología molecular, como, por ejemplo, estudios de carga viral. El desarrollo más amplio de estos aspectos se puede encontrar en el procedimiento microbiológico de la Sociedad Española de Enfermedades Infecciosas y Microbiología Clínica (SEIMC) número 32: Recomendaciones para la implantación de la normativa de calidad ISO 15189 en el Laboratorio de Microbiología Clínica: bacteriología y serología (2.a edición, 2009) (Disponible en: URL: www.seimc.org/protocolos/microbiologia).
Definición de alcancesEl alcance se define como el conjunto de estudios microbiológicos para los que el laboratorio está acreditado o va a solicitar la acreditación. Para cada estudio debe incluir el tipo de muestra, la determinación o análisis que se va a realizar y el método que se va a emplear.
En la tabla 1 se definen los contenidos mínimos para los estudios bacteriológicos más frecuentes. Algunos son fáciles de delimitar y se consideran obligados, mientras que otros pueden considerarse optativos y podrían ser motivo de opinión entre los profesionales.
Contenidos mínimos para los estudios bacteriológicos más frecuentesa
| Muestras | Estudios obligados | Estudios optativosb |
| Orinas |
|
|
| Heces |
|
|
| Muestras respiratorias | ||
| Exudado faríngeo |
|
|
| Exudado nasofaríngeo |
|
|
| Exudado nasal (portador de Staphylococcus aureus) |
|
|
| Muestras de las vías respiratorias inferiores: esputo, aspirado traqueal, broncoaspirado (BAS), LBA, cepillado bronquial |
|
|
| Exudado ótico |
|
|
| Muestras genitalesc | ||
| Exudado vaginal (incluye patógenos fúngicos y parasitarios) |
|
|
| Exudado de cuello uterino (endocervical) |
|
|
| Exudado uretral |
|
|
| Semen (diagnóstico de prostatitisd y estudio de infertilidad) |
|
|
| Exudado rectal |
|
|
| Líquido cefalorraquídeo (LCR) |
|
|
| Hemocultivos |
|
|
| Puntas de catéteres intravasculares |
|
|
| Líquidos biológicos normalmente estériles (ascítico, peritoneal, pleural, articular, etc.) |
|
|
| Exudado conjuntival |
|
|
| Miscelánea: abscesos, colecciones purulentas, úlceras, exudados de heridas superficiales y profundas, exudados umbilicales, otros exudados |
|
|
ITS: infección de transmisión sexual.
Aunque este documento se centra en los estudios bacteriológicos, el estudio microbiológico de ciertas muestras carece de sentido si no se realiza un enfoque de los diferentes patógenos que pueden estar implicados (fúngicos, parasitarios o víricos).
En relación con los estudios serológicos, la definición de los alcances debe incluir los siguientes elementos:
- a)
El tipo de muestra, que mayoritariamente es suero humano, aunque algunos de los ensayos son, además, de aplicación a plasma. También se pueden aplicar a otras muestras, como líquido cefalorraquídeo para el estudio de infecciones del sistema nervioso central, muestras de saliva para el diagnóstico o el establecimiento del estado inmunitario en determinadas situaciones y, por último, la sangre seca sobre papel de filtro, especialmente útil en el diagnóstico retrospectivo de infecciones congénitas en lactantes, empleando la muestra tomada al nacimiento para el cribado habitual de metabolopatías congénitas.
- b)
El analito objeto del ensayo, que cuando se pretenda establecer el estado inmunitario serán anticuerpos totales (IgG+IgM+IgA) o IgG, y si el fin es diagnóstico, anticuerpos de clase IgM.
- c)
La metodología empleada. En general, las técnicas en fase sólida (ELISA o IF) son de aplicación a la detección de anticuerpos específicos de clase IgG, IgM o IgA, en tanto que las técnicas en fase líquida (neutralización, fijación del complemento) no identifican el isotipo de la respuesta de los anticuerpos específicos detectados.
El laboratorio debe disponer de documentos donde se describan los procesos de tipo organizativo o técnico necesarios para implantar la norma UNE-EN-ISO 15189:2007. Es importante establecer un sistema para controlar los documentos, de manera que las versiones actualizadas se encuentren disponibles en los puntos de uso, y se evite el uso de documentos no válidos u obsoletos2. La documentación de calidad debe incluir, entre otros, los documentos que se indican a continuación:
- a)
Manual de calidad5. Describe el SGC del laboratorio y sus actividades, haciendo referencia a los documentos donde se desarrollan los procedimientos. Ha de incluir pautas de organización y gestión (política y objetivos de calidad, funciones y responsabilidades de los distintos puestos de trabajo, gestión de la información, compras, sistemas de mejora continua, etc.) y pautas de tipo técnico (requisitos de las fases preanalítica, analítica y postanalítica del estudio microbiológico, control de equipos e instalaciones, aseguramiento de la calidad, entre otros).
- b)
Manual de extracción y transporte de muestras3. Incluye información sobre las muestras clínicas y estudios recomendados según el síndrome clínico o infeccioso, instrucciones para la recogida, transporte y conservación de las muestras, criterios de aceptación y rechazo, así como pruebas disponibles, posibles resultados y orientación para la interpretación de éstos.
- c)
Procedimientos de gestión o procedimientos generales. Describen la sistemática para realizar procedimientos de tipo organizativo; algunos ejemplos son el de control de la documentación, el de gestión de compras, equipos, personal, bioseguridad.
- d)
Procedimientos normalizados de trabajo (PNT). Contienen las instrucciones detalladas para la realización de un análisis microbiológico o procedimiento técnico.
- e)
Registros5. Documentos que proporcionan evidencias de actividades efectuadas o de resultados obtenidos, como las hojas de trabajo, los informes de resultados, los informes de control de calidad, las actas de reuniones, y otros. ENAC recomienda archivar durante 5 años los registros de control de calidad y los informes de laboratorio; para otros recomienda un mínimo de 3 años6.
- f)
Formularios. Impresos o fichas en formato electrónico con espacios en blanco, que una vez cumplimentados se convierten en registros.
El análisis microbiológico consta de 3 fases: preanalítica (petición de la prueba, recogida y transporte de la muestra y recepción y registro de la muestra en el laboratorio), analítica (preparación y realización de la prueba y obtención de los resultados) y postanalítica (elaboración del informe de resultados, validación y distribución de los resultados)3. La norma UNE-EN-ISO 15189:20072, entre otros, establece los siguientes requisitos:
- 1.
Fase preanalítica. Es necesario elaborar un manual de extracción y transporte de muestras y controlar que está disponible y actualizado en los puntos de extracción de muestras3. Además, es necesario que el laboratorio disponga de un documento que describa la sistemática de trabajo en el área de recepción de muestras y la normativa sobre la aceptación y el rechazo de las muestras2,6.
- 2.
Fase analítica. El laboratorio debe utilizar métodos validados y descritos en los PNT, en los que se aportará la siguiente información:
- •
Propósito y alcance. Especificaciones sobre el estudio que se va a realizar, a partir de qué muestras y con qué objetivo.
- •
Principio del procedimiento. Fundamento científico de la metodología empleada.
- •
Documentos de consulta. Manuales y guías en los que se basa el procedimiento.
- •
Muestra. Tipos de muestra para los que es aplicable el procedimiento, método de obtención, volumen necesario, contenedor adecuado, condiciones de conservación y criterios de rechazo aplicables.
- •
Equipos y reactivos. Medios de cultivo, reactivos, material fungible, estufas, analizadores y cualquier equipo necesario para la realización de la técnica.
- •
Desarrollo del ensayo. Protocolo claro y pormenorizado, que permita que el personal pueda realizarlo con la mínima variabilidad posible.
- •
Obtención de resultados. En bacteriología deben quedar definidos los criterios para obtener e interpretar los resultados. En las técnicas cualitativas se hará una relación de los posibles resultados y en las técnicas cuantitativas o semicuantitativas se describirán los cálculos necesarios para obtenerlos, especificando las unidades de medida. En serología hay que definir el criterio usado para calcular los resultados, es de especial interés la definición del valor de corte, así como de la zona gris (resultados indeterminados).
- •
Interpretación de resultados. Es necesario predefinir los comentarios que pueden acompañar a los informes para contribuir a la interpretación de éstos.
- •
Control de calidad. En bacteriología, cuando sea posible, se podrían incluir controles internos con una periodicidad establecida. En las técnicas comerciales es conveniente utilizar de manera reglada los controles que proporcione el kit. En los ensayos serológicos, los kits comerciales deben incluir controles suficientes para asegurar su correcto funcionamiento. Además, se podrían incluir controles propios del laboratorio para valorar la precisión interensayo e intraensayo del método.
- •
Fuentes de interferencias. En los estudios bacteriológicos, la presencia de microorganismos saprofitos, el contacto con antisépticos o desinfectantes y el tratamiento antimicrobiano previo, pueden alterar los resultados. En serología las principales fuentes de interferencias son la contaminación, la lipemia o la hemólisis de la muestra, además de las reacciones cruzadas que existen entre los diferentes patógenos7 y la presencia del factor reumatoide en determinaciones de IgM realizadas con técnicas indirectas.
- •
Posibles fuentes de variabilidad. En estudios en los que la lectura es visual puede influir la experiencia y pericia del observador. En los ensayos que tienen fases de lavado (tinciones o técnicas serológicas en fase sólida), un lavado insuficiente puede proporcionar resultados erróneos o difícilmente interpretables. Algo similar ocurre con el material volumétrico, fundamentalmente el empleado para dispensar pequeños volúmenes; se necesitan sistemas precisos al máximo.
- •
Responsabilidades. Actividades que debe realizar o controlar el personal que participa en el procedimiento.
- •
Precauciones de seguridad. Recomendaciones sobre las buenas prácticas de laboratorio teniendo en cuenta especialmente el carácter potencialmente infeccioso de las muestras clínicas y de los reactivos empleados en algunos estudios.
- •
Bibliografía. Referencias bibliográficas actualizadas que sirven de fundamento al procedimiento.
- •
- 3.
Fase postanalítica. Una vez revisados y validados los resultados obtenidos se genera el informe, que debe incluir toda la información necesaria para su correcta interpretación por el clínico. Se puede distribuir en papel o en formato electrónico, siempre que el laboratorio cumpla los requisitos legales y garantice la confidencialidad de los resultados. La información telefónica de los resultados debe estar regulada y quedar registrada, así como la modificación de los informes.
El laboratorio debe disponer de un programa documentado de control de calidad interno en el que se detallen las actividades que se van a realizar, la frecuencia de éstas y las responsabilidades del personal en este tema8. Cuando en un procedimiento de control de calidad se detecte algún resultado anómalo hay que investigar las causas y una vez halladas llevar a cabo las acciones correctoras necesarias para eliminar la fuente de error y tomar medidas preventivas para que no vuelva a ocurrir9. Las actividades de control de calidad y sus resultados, así como las posibles acciones correctivas realizadas, deben registrarse indicando la persona que las ha llevado a cabo.
BacteriologíaEs necesario mantener una colección de cepas de referencia para su uso en los procedimientos de control de calidad interno. El mantenimiento de estas cepas en el laboratorio debe garantizar la conservación de su pureza y características3,8,10.
- a)
Medios de cultivo. Si los medios son comerciales, deben proporcionarlos fabricantes cetificados. Antes de utilizarlos hay que comprobar las características físicas (color, espesor del medio, precipitados, placas rotas, excesivo número de burbujas, etc.) y las posibles contaminaciones. Existen medios denominados exentos que no requieren que en el laboratorio se compruebe su funcionamiento antes de ponerlos en uso, siempre que el fabricante aporte las especificaciones de calidad correspondientes11. En los medios no exentos y en los exentos que demuestren alguna deficiencia sí es necesario comprobar que permiten el crecimiento de microorganismos específicos y/o inhiben el crecimiento de otros y que producen una adecuada respuesta bioquímica o morfologías típicas de los microorganismos ensayados. Cuando los medios de cultivo se preparan en el propio laboratorio hay que controlar todos los componentes, el procedimiento de elaboración, el envasado, etiquetado y almacenamiento y, por último, comprobar las características físicas, la esterilidad y el funcionamiento3,8,12. En el documento de la SEIMC “Recomendaciones generales para el control de calidad interno en microbiología clínica” se recogen aspectos relacionados con los requisitos y con las frecuencias del control de calidad de los medios de cultivo10.
- b)
Reactivos. Deben estar etiquetados con su contenido, concentración, condiciones de conservación, fecha de preparación o de reconstitución y fecha de caducidad o período recomendado de almacenamiento. Debe existir un documento para cada reactivo o kit en el que se detallen sus componentes y su proporción, el ensayo microbiológico para el que se utiliza, el método de lectura de los resultados, los controles que deben utilizarse, los criterios de aceptabilidad y la frecuencia de control. En el caso de los reactivos comerciales, el fabricante aportará evidencias del cumplimiento de los criterios de calidad. Para los reactivos preparados en el laboratorio debe existir una ficha técnica de elaboración que incluya el control del funcionamiento adecuado y debe quedar registrado el proceso de elaboración y quién lo ha llevado a cabo3,8,10.
- c)
Antibiograma. El control de calidad del antibiograma se basa en la realización de estas pruebas en condiciones estándares13 y con cepas de referencia genéticamente estables. Los medios utilizados en el antibiograma son no exentos11, por lo tanto, es necesario controlar cada nuevo lote; igualmente ocurre con cada nuevo lote de discos, tiras y antibióticos que se reciba en el laboratorio. La densidad del inóculo también afecta al rendimiento del antibiograma, por lo que es recomendable controlar periódicamente estos inóculos realizando un cultivo cuantitativo de la suspensión bacteriana empleada y comprobando que el recuento obtenido es correcto, según el microorganismo ensayado y la técnica empleada (normalmente entre 105–108UFC/ml)13. La lectura interpretada del antibiograma es una extraordinaria herramienta para el control de la calidad del antibiograma, ya que permite detectar resultados atípicos.
- d)
Sistemas automatizados de identificación y antibiograma. Es necesario realizar una validación inicial o comprobación del funcionamiento con cepas de referencia cuyos perfiles de sensibilidad son conocidos y, posteriormente, se podrá optar por realizar controles con una periodicidad fija o para cada nuevo lote de reactivos. Si cambia el procedimiento, el equipo o el tipo de paneles utilizados, es necesario realizar una nueva validación. Estas actividades deben quedar registradas de modo que pueda verificarse fácilmente su trazabilidad.
- e)
Sistemas automatizados de hemocultivos. A la puesta en marcha del equipo se debe llevar a cabo una validación inicial del sistema de detección de crecimiento microbiano (sensores) que incluyen estos equipos, utilizando no solo los microorganismos aislados habitualmente, sino también los de lento crecimiento y los microorganismos exigentes.
Los medios de hemocultivo comerciales están exentos11 del control de calidad siempre que el fabricante aporte evidencias del control realizado. Sin embargo, sí se debe confirmar inicialmente su capacidad para permitir el crecimiento de microorganismos exigentes.
El laboratorio de diagnóstico serológico debe establecer un sistema de aseguramiento de la calidad que incluya aspectos preanalíticos, analíticos y postanalíticos. Al igual que sucede en otro tipo de laboratorios, la mayoría de los errores ocurren en las fases preanalítica y postanalítica14. En la fase preanalítica hay que considerar factores como la correcta identificación y la calidad de la muestra (ausencia de hemólisis, contaminación o lipemia). Es recomendable utilizar sistemas automáticos para el reconocimiento de las muestras, para evitar errores en la identificación. En la fase postanalítica hay que controlar los posibles errores en la transcripción de los resultados, el tiempo de respuesta y, muy importante, la interpretación de los resultados, que siempre debe realizarla personal especializado.
En lo que se refiere a la fase analítica, se deben emplear siempre ensayos validados2,6 (se ha confirmado a través de evidencias objetivas que cumplen los requisitos previstos). En las técnicas comerciales con marcado CE, el fabricante es el responsable de la validación; no obstante, cuando un ensayo se incorpora a la rutina o cuando se produce un cambio en éste es recomendable realizar una verificación in situ determinando la exactitud y la precisión intraensayo e interensayo y en los ensayos cuantitativos, además debe establecerse la linealidad. Cuando se utilicen métodos desarrollados en el laboratorio o modificaciones sobre los métodos comerciales, o bien se vayan a aplicar para un fin distinto al establecido por el fabricante, es necesario hacer una validación en el propio laboratorio, determinando exactitud, precisión, sensibilidad y especificidad y, en los ensayos cuantitativos, la linealidad15.
Para comprobar el funcionamiento de un ensayo se deben incluir sueros de control positivo (uno o varios) y suero de control negativo. Igualmente, se deben incluir, en lo posible, controles de todos los componentes de la reacción que sean precisos.
Los ensayos comerciales que no permiten manipulación por el operador, realizados en sistemas cerrados, suelen precisar calibración, que se debe realizar al incorporar un nuevo lote, periódicamente, o al inicio de cada sesión de trabajo; además es necesario incluir sueros control. Otro tipo de ensayos comerciales (ELISA, IFI, aglutinación y otros), aunque pueden realizarse de forma manual o de forma automática, tienen un alto componente de manipulación por el operador, por lo que incluyen controles para asegurar que cada ensayo ha funcionado correctamente. En general incluyen un suero negativo y uno o varios controles positivos, alguno de los cuales se emplea como calibrador. Los valores obtenidos por los sueros de control siempre deben estar en el rango esperado, establecido por el fabricante. En los ensayos en fase sólida (ELISA o IFI) se deben incluir pocillos de blanco, para excluir la unión inespecífica del anticuerpo secundario a la fase sólida, y controlar la obtención de los resultados positivos falsos generalizados. Por otra parte, los ensayos comerciales modificados para una aplicación diferente a la especificada por el fabricante, requieren la inclusión de controles específicos de la nueva aplicación del ensayo.
Aseguramiento de la calidad. Control de calidad externo: intercomparacionesLa participación en programas de evaluación externa de la calidad es un requisito fundamental, su fin es contrastar los resultados de un laboratorio con otros laboratorios2,3,8. Estos programas pueden estar organizados nacional o internacionalmente y deben estar reconocidos por los servicios de salud, las sociedades científicas u otros organismos. Es recomendable analizar las muestras del programa dentro de la rutina del laboratorio como si fueran muestras clínicas, utilizando las mismas técnicas y realizadas por el personal que intervendría en el procesamiento de ese tipo de muestras. Los resultados emitidos por el organismo organizador deben revisarse dejando constancia de las conclusiones obtenidas. Si el resultado obtenido no es el adecuado habrá que estudiar las causas y poner en marcha acciones correctivas.
La SEIMC dispone de un programa de evaluación externa de la calidad muy completo; sin embargo, el número de controles realizados en alguna de las áreas es bajo. Los programas del Colegio de Patólogos Americanos (URL: www.cap.org) y del Reino Unido (United Kingdon National External Quality Assessment Schemes [URL: www.ukneqas.org.uk/]) pueden complementarlo. También existe la posibilidad de organizar un sistema de intercomparaciones entre varios laboratorios sin la participación de ninguna entidad organizadora. Para esto, un grupo de laboratorios interesados y con la periodicidad pactada se van turnando en la preparación y en la distribución de las muestras objeto de la intercomparación.
Control de equiposLa calidad de un análisis microbiológico depende, entre otros factores, del buen uso y funcionamiento de los equipos. El laboratorio debe contar con los equipos necesarios para la correcta ejecución de los ensayos y disponer de un inventario actualizado de los equipos en uso. Cuando se recibe un equipo en el laboratorio hay que comprobar que cumple con las especificaciones previstas, además de verificar la documentación que lo acompañe y su estado. Antes de ponerlo en uso se realizará una validación inicial o confirmación a través de evidencias objetivas de que los requerimientos para el uso o aplicación propuestos se cumplen5; en el caso de los instrumentos de medida, la validación puede incluir su caracterización determinando la incertidumbre (parámetro que permite conocer el posible error en las medidas)2,16 y otras características, como la exactitud o la precisión. En general, en los laboratorios de microbiología no es necesario el cálculo de la incertidumbre, a no ser que el organismo acreditador o el laboratorio considere crítica una medida en un determinado equipo. La documentación necesaria para cada equipo es la siguiente:
- a)
Ficha del equipo con la descripción y el código del equipo, la localización habitual, los datos del fabricante, la fecha de recepción y de puesta en servicio, el procedimiento de validación, verificación, mantenimiento y su periodicidad y precauciones de bioseguridad.
- b)
Manual del equipo, que junto con las instrucciones de uso y mantenimiento, deben estar disponibles para el personal del laboratorio autorizado.
- c)
Registros de cada una de las revisiones realizadas.
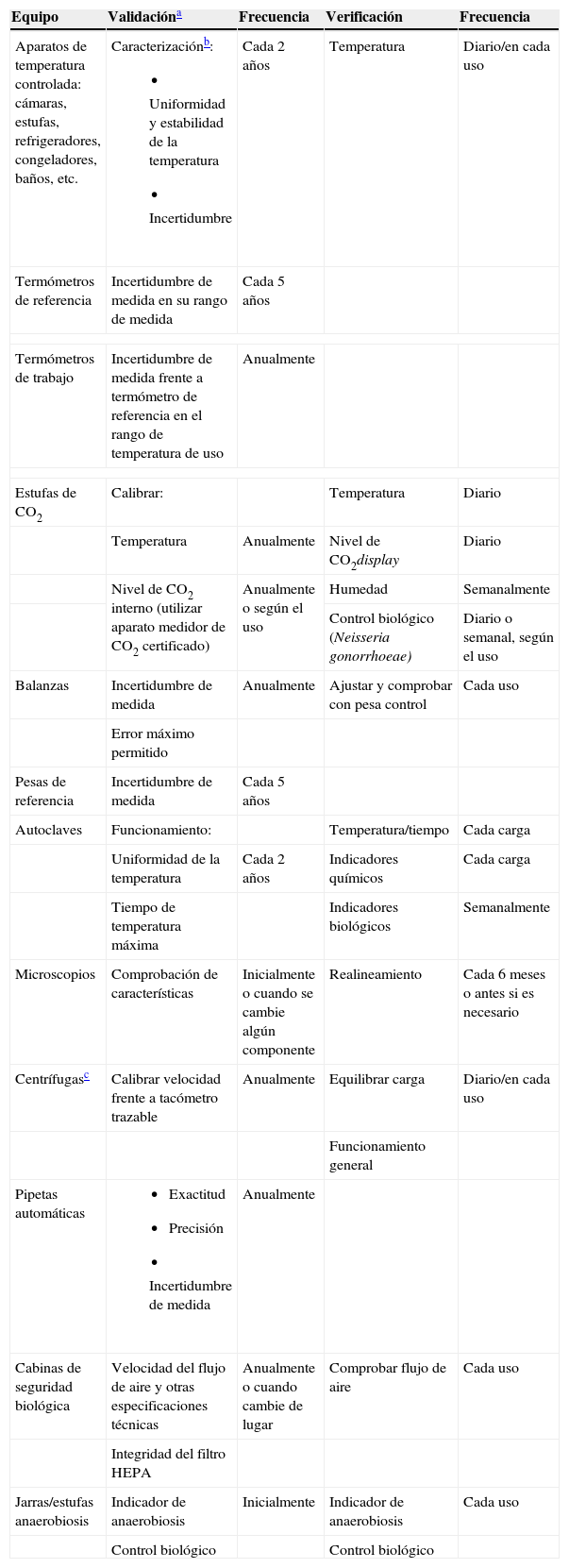
La norma UNE-EN-ISO 15189:20072 exige que el laboratorio disponga de un programa de validación, verificación y mantenimiento para los equipos que puedan influir significativamente en los resultados de los ensayos. Cada procedimiento debe quedar registrado y los registros deben mantenerse y estar fácilmente disponibles durante la vida útil del equipo. Después de la validación inicial, los períodos de validación o verificación5 (comprobación en el día a día que un equipo cumple con sus especificaciones) se establecen en función de la naturaleza del equipo, condiciones de uso, historia previa del equipo y recomendaciones del fabricante. En la tabla 2 se indica la frecuencia recomendada para algunos equipos usados en el laboratorio de microbiología.
Tabla 2.Recomendaciones para la validación y verificación de equipos
Equipo Validacióna Frecuencia Verificación Frecuencia Aparatos de temperatura controlada: cámaras, estufas, refrigeradores, congeladores, baños, etc. Caracterizaciónb: - •
Uniformidad y estabilidad de la temperatura
- •
Incertidumbre
Cada 2 años Temperatura Diario/en cada uso Termómetros de referencia Incertidumbre de medida en su rango de medida Cada 5 años Termómetros de trabajo Incertidumbre de medida frente a termómetro de referencia en el rango de temperatura de uso Anualmente Estufas de CO2 Calibrar: Temperatura Diario Temperatura Anualmente Nivel de CO2display Diario Nivel de CO2 interno (utilizar aparato medidor de CO2 certificado) Anualmente o según el uso Humedad Semanalmente Control biológico (Neisseria gonorrhoeae) Diario o semanal, según el uso Balanzas Incertidumbre de medida Anualmente Ajustar y comprobar con pesa control Cada uso Error máximo permitido Pesas de referencia Incertidumbre de medida Cada 5 años Autoclaves Funcionamiento: Temperatura/tiempo Cada carga Uniformidad de la temperatura Cada 2 años Indicadores químicos Cada carga Tiempo de temperatura máxima Indicadores biológicos Semanalmente Microscopios Comprobación de características Inicialmente o cuando se cambie algún componente Realineamiento Cada 6 meses o antes si es necesario Centrífugasc Calibrar velocidad frente a tacómetro trazable Anualmente Equilibrar carga Diario/en cada uso Funcionamiento general Pipetas automáticas - •
Exactitud
- •
Precisión
- •
Incertidumbre de medida
Anualmente Cabinas de seguridad biológica Velocidad del flujo de aire y otras especificaciones técnicas Anualmente o cuando cambie de lugar Comprobar flujo de aire Cada uso Integridad del filtro HEPA Jarras/estufas anaerobiosis Indicador de anaerobiosis Inicialmente Indicador de anaerobiosis Cada uso Control biológico Control biológico aLa norma UNE-EN-ISO 15189:2007 y la documentación de ENAC relacionada se refieren, en algunos casos, a estos procedimientos como calibración. Además de la frecuencia indicada para la validación se realizará siempre inicialmente antes de la puesta en uso del equipo y después de cada reparación o modificación importantes.
El mantenimiento permite que un equipo esté en condiciones de uso adecuadas y puede ser de tipo correctivo o preventivo. El mantenimiento en los equipos como baños termostáticos, estufas, refrigeradores, cabinas de seguridad biológica y microscopios consiste en la limpieza, la inspección de posibles daños y la comprobación general. El documento G–ENAC-4. Rev. 317 establece una frecuencia orientativa para estas operaciones según el equipo. En el caso de los sistemas automatizados debe existir un programa de mantenimiento preventivo que siga las recomendaciones del fabricante8. Cuando el resultado de la revisión de un equipo no es satisfactorio, se estudiarán las causas y la repercusión en los ensayos realizados, estableciéndose las acciones correctoras pertinentes.
- •
El laboratorio de microbiología debe disponer de una relación detallada de los puestos de trabajo, en la que se defina para cada puesto la titulación, la cualificación, las funciones y las responsabilidades del personal que lo ocupa. Además de poseer la titulación requerida, el personal del laboratorio debe cualificarse y ser competente5 para desarrollar las tareas específicas de su puesto de trabajo; para esto, debe conocer los manuales o procedimientos del laboratorio y someterse a un entrenamiento adecuado en la actividad que va a realizar. Posteriormente, es necesario comprobar que el personal sigue siendo competente para desarrollar su labor correctamente; para esto se debe evaluar la competencia del personal anualmente, dejando evidencias de esta evaluación18,19. El laboratorio debe mantener registros de la formación y cualificación, de la experiencia profesional y de la competencia de todo el personal6.
Por último, es necesario establecer un sistema para detectar las necesidades de formación del personal y elaborar un plan deformación2 que se puede revisar anualmente. Para desarrollarlo se pueden organizar sesiones docentes (revisiones de temas, bibliografía, sesiones clínicas y otros) de las que quedará constancia mediante un acta. También es recomendable la asistencia a congresos, reuniones, cursos o simposios que proporcionen actualización científica y técnica.
Sistemas de informaciónEl laboratorio debe disponer de un sistema de información que garantice la integridad y la confidencialidad de los datos del paciente y de los resultados generados en el procesamiento de las muestras microbiológicas20. Es necesario elaborar un procedimiento general de gestión de la información que incluya:
- a)
La política de utilización del sistema informático por el personal del laboratorio, definiendo los distintos niveles de autorización de los usuarios, así como la metodología para el registro de peticiones, introducción, revisión y validación de resultados, impresión, autorización y distribución de informes.
- b)
La descripción del sistema de almacenamiento de datos (en formato papel o electrónico), que debe permitir su fácil recuperación e impedir la pérdida de datos relevantes, el acceso no autorizado o una manipulación indebida.
- c)
El formato del informe de resultados y su contenido mínimo, tipos de informes (de rutina, previos, urgentes, procedentes de laboratorios externos), métodos para comunicar los resultados (por teléfono, vía electrónica, otros), así como para modificar informes ya emitidos.
- d)
Metodología establecida para garantizar la trazabilidad5, de manera que sea posible identificar a los individuos que han participado en cada uno de los pasos del proceso analítico desde que la muestra se recibe en el laboratorio hasta que el resultado se envía al facultativo que solicitó el estudio. En el caso de existir consultas de los resultados por vía electrónica, el acceso se hará a través de una clave personal, de manera que impida la manipulación o modificación de los datos y el proceso será trazable, siendo posible conocer quién y cuándo ha accedido al sistema y qué ha consultado.
- e)
Por último, las responsabilidades del personal en cuanto a la gestión de la información.
El laboratorio debe definir en un procedimiento general su sistemática para la gestión de pedidos, estableciendo los requisitos que se van a exigir a proveedores y productos, así como la sistemática para solicitar y recibir pedidos. La norma UNE-EN-ISO 15189:20072 también recoge la obligatoriedad de definir un sistema para el control de inventario de los productos. El laboratorio debe evaluar a sus proveedores teniendo en cuenta el cumplimiento de los requisitos técnicos establecidos, plazos de entrega adecuados, asesoramiento posventa, garantía de calidad o rapidez de respuesta ante problemas puntuales. Los proveedores aprobados se reevaluarán periódicamente para asegurar que mantienen la calidad de sus servicios.
Análisis realizados por laboratorios externosEl laboratorio de microbiología debe asegurarse de la competencia y calidad de los laboratorios a los que envía muestras para su estudio de manera similar a la que se realiza con los proveedores. Es recomendable que el laboratorio contratado para realizar análisis incluidos en el alcance de acreditación esté acreditado por ENAC o por cualquier organismo de acreditación con el que ENAC haya firmado un acuerdo de reconocimiento European Cooperation for Accreditation [EA], International Laboratories Accreditation Cooperation [ILAC ], o en su defecto, que tenga certificado su sistema de calidad6. Hay que disponer de un registro de las muestras enviadas y de los laboratorios a los que se han enviado cada una de éstas y archivar un duplicado de los resultados recibidos. El laboratorio puede reenviar directamente el informe remitido por el laboratorio subcontratado o bien puede preparar un informe propio que incluya los resultados exactos contenidos en el informe original. Aquellos resultados obtenidos de laboratorios externos no acreditados se informarán como no acreditados por la ENAC.
Mejora continua: sistemas de evaluaciónEl laboratorio debe monitorizar de manera continua el funcionamiento del SGC para corregir las posibles desviaciones o errores detectados, que se pueden clasificar como:
- a)
Incidencias: pueden influir de manera negativa, aunque no significativamente, sobre los servicios prestados por el laboratorio, pero no producen alteración en los resultados de los estudios.
- b)
No conformidades5: pueden afectar significativamente a la calidad de los procedimientos y pueden alterar los resultados de los estudios microbiológicos.
Cuando se detecta una desviación hay que determinar las causas que la han provocado, así como la extensión y las consecuencias del problema y establecer acciones correctivas para eliminar estas causas y evitar que la desviación se repita9. Es necesario realizar un seguimiento de las medidas correctoras, evaluando la eficacia de éstas y comprobando la resolución del problema. Todo el procedimiento debe quedar registrado.
Una importante herramienta de mejora continua son las auditorías internas2,5,6, que tienen como objetivo verificar la implantación del SGC en el laboratorio, comprobando que se cumplen los requisitos técnicos y de gestión establecidos y que la documentación relacionada se mantiene actualizada. Se llevan a cabo de manera sistemática, generalmente con periodicidad anual, por personal del mismo laboratorio independiente de la actividad auditada o por auditores externos. En el Informe de Auditoría Interna se reflejan las desviaciones detectadas, frente a las que se establecerá un plan de acciones correctoras que deben documentarse y realizarse en un plazo acordado.
Otra herramienta para la mejora continua son las reuniones de Revisión por la Dirección del Laboratorio2 (director del laboratorio, jefes de sección u otros facultativos, responsable de calidad, supervisor/a del laboratorio) que deben realizarse al menos una vez al año para tratar temas relacionados con la calidad, como el resultado de la auditoría interna, el tratamiento de las desviaciones detectadas, la revisión y el seguimiento de objetivos e indicadores de calidad, los resultados de evaluaciones externas de calidad, la revisión de los recursos humanos disponibles, la evaluación de proveedores y cualquier otro aspecto de interés.
Por último, es necesario establecer indicadores de calidad3,8 para monitorizar de manera sistemática las fases preanalítica, analítica y postanalítica del estudio microbiológico y detectar oportunidades de mejora. En la construcción de un indicador hay que tener en cuenta el objetivo que se pretende conseguir, los estándares o valores de control que se van a considerar aceptables, la fuente de datos y, por último, la periodicidad de las mediciones, que dependerá de la gravedad de las posibles desviaciones detectadas.

