




La evolución del tratamiento antirretroviral (TAR) en los últimos años ha permitido que se disponga de opciones seguras, eficaces a corto y a largo plazo, y también sencillas hasta el punto de que la mayoría de las combinaciones se administran una vez al día, se concentran en uno o 2 comprimidos, y apenas existen fracasos terapéuticos. Pese a todo, continúan existiendo motivos para el cambio de un TAR eficaz en pacientes inmunodeprimidos, cambio que siempre se debe realizar sin perder la eficacia virológica. Con el aumento de la supervivencia en los últimos años y la necesidad de tratamiento de por vida, es importante disponer de pautas simples y seguras para mejorar la calidad de vida de los pacientes con VIH. El ensayo clínico EMERALD demuestra la no inferioridad del cambio a Symtuza®, combinación de darunavir, cobicistat, em-tricitabina y tenofovir alafenamida en un solo comprimido, en pacientes con supresión viral mantenida. Información sobre el suplemento: este artículo forma parte del suplemento titulado «Darunavir, cobicistat, emtricitabina y tenofovir alafenamida coformulados en el tratamiento de la infección por el VIH», que ha sido patrocinado por Janssen.
Because of the development of antiretroviral therapy (ART) in the last few years, we now have safe, effective short- and long-term options and also simple regimens, with most combinations being administered once daily in one or 2 tablets and almost no treatment failures. Nevertheless, there are still reasons to switch an effective ART in patients who have been suppressed, a switch that must always be made without losing virological efficacy. With longer survival and the need for lifelong treatment, it is important to have simple and safe treatment options to improve the quality of life in patients with HIV. The EMERALD clinical trial demonstrates the non-inferiority of the switch to Symtuza®, a combination of darunavir, cobicistat, emtricitabine and tenofovir alafenamide in a single tablet, in patients with sustained viral suppression. Supplement information: This article is part of a supplement entitled “Co-formulated cobicistat-boosted darunavir, emtricitabine, and tenofovir alafenamide for the treatment of HIV infection”, which is sponsored by Janssen.
La generalización del tratamiento antirretroviral (TAR) combinado ha sido responsable de la disminución de la morbimortali-dad asociada con el VIH1-5. Además, en los últimos años se han objetivado otros beneficios adicionales: disminución de la incidencia de otras comorbilidades6 y prevención de la transmisión7,8, lo que en algunas áreas geográficas se ha traducido en una disminución de la incidencia del VIH2. Por estos motivos, desde mediados de 2015 en todas las guías se recomienda administrar TAR a todas las personas con VIH, independientemente de la cifra de linfocitos CD49,10.
Todo esto ha sido posible gracias a la evolución de los fármacos antirretrovirales, no solo desde el punto de vista de la eficacia sino también de la seguridad y la conveniencia. Disponemos de fármacos eficaces y seguros que permiten mantener indetectable la carga viral plasmática en la mayoría de los pacientes, independientemente de la línea de tratamiento que estén tomando10. En los ensayos clínicos fundamentales de los fármacos comercializados más recientemente existen tasas muy bajas de suspensión de tratamiento por efectos adversos, hecho que se reproduce en la práctica clínica diaria10,11. La aparición de los nuevos inhibidores de la integrasa o de tenofovir alafenamida (TAF) son un claro ejemplo de ello11,12. Además, se han desarrollado coformulaciones que permiten un TAR completo con un comprimido diario (STR, single tablet regimen) o, al menos, pautas administradas una sola vez al día, con lo que se puede mejorar la adherencia, la calidad de vida y los resultados inmunovirológicos11-14. Symtuza® es una de las coformulaciones comercializadas más recientemente y la primera que incluye en un solo comprimido emtricita-bina (FTC), TAF y un inhibidor de la proteasa: darunavir (DRV), potenciado con cobicistat (c)11,14,15.
En este escenario podríamos pensar que el cambio de tratamiento en pacientes con carga viral (CV) indetectable no tiene sentido, pero los avances conseguidos y un mejor conocimiento de las resistencias a los antirretrovirales pueden hacernos reconsiderar las pautas que estamos administrando a nuestros pacientes y plantearnos otras opciones terapéuticas alternativas y eficaces que pueden ser más adecuadas en distintas situaciones. Los motivos de cambio también han estado influidos por las opciones terapéuticas disponibles en cada momento y la evidencia científica disponible. Los aspectos relacionados con el cambio de tratamiento aparecen recogidos en las distintas guías terapéuticas. Está claro que, siempre que se plantee un cambio en un paciente que se encuentra indetectable, el objetivo debe ser mantener la carga viral suprimida sin comprometer futuras opciones10,16-18.
En este artículo vamos a revisar los aspectos relacionados con el cambio de TAR en un paciente con VIH y carga viral indetectable, y la información disponible del ensayo clínico EMERALD14-15, en el cual se estudia la eficacia y seguridad del cambio a Symtuza® en pacientes estables en tratamiento con un inhibidor de la proteasa (IP) potenciado con tenofovir disoproxil (TDF) y FTC frente al hecho de continuar con la misma pauta.
Cambios del tratamiento antirretroviral en el paciente con carga viral undetectable o suprimidaEl primer concepto que debe quedar claro es lo que se considera carga viral suprimida en el momento de realizar un cambio de tratamiento. En los ensayos clínicos de estrategias de cambio se define como carga viral inferior a 50 copias/ml confirmada, al menos, durante 6 meses. Como norma general, cuanto más prolongado sea el período de supresión virológica es menos probable que el cambio de TAR se asocie con un fracaso virológico. El cambio puede ser proactivo cuando se realiza de manera preventiva o reactivo cuando el tratamiento que está tomando deja de ser el ideal para el paciente por algún motivo10,16-18.
Motivos de cambio y objetivos del nuevo tratamientoExisten muchos motivos para cambiar un TAR eficaz: intolerancia, toxicidad documentada o prevención de esta a largo plazo, nuevas comorbilidades o cambios en las existentes previamente, interacciones farmacológicas, simplificación (ya sea disminución del número de comprimidos o de dosis diarias), requerimientos dietéticos, embarazo o disminución del coste del tratamiento10,18.
El objetivo del cambio de TAR eficaz es mantener la supresión virológica y optimizar el TAR de acuerdo con las características y las preferencias del paciente, para eliminar o mejorar efectos adversos, facilitar el tratamiento adecuado de las comorbilidades y mejorar su calidad de vida. El especialista clínico debe revisar periódicamente los posibles efectos adversos o la tolerabilidad del TAR. No debería asumirse que la persona con VIH está bien adaptada y tolera bien el régimen de TAR por el mero hecho de que la CV esté suprimida. En ocasiones, la adherencia se consigue gracias a un sobreesfuerzo del paciente, que es capaz de sobrellevar efectos adversos que pueden ser erróneamente entendidos como inevitables10,16-18.
Estrategias de cambioEl cambio proactivo es obligado cuando evidencias sólidas avalan que el paciente corre más riesgo de presentar un efecto adverso grave o irrecuperable si se mantiene el TAR actual que si se cambia. El ejemplo clásico es la lipoatrofia10.
El cambio reactivo es obligado si el efecto adverso va a desaparecer tras el cambio de TAR, como los efectos adversos del sistema nervioso central causados por efavirenz10,18.
Una vez que se ha tomado la decisión de cambiar el tratamiento, hay que revisar de forma exhaustiva la historia clínica del paciente, incluyendo la historia del TAR junto con la de las CV de VIH, motivos de cambio y problemas de tolerabilidad, así como la de fracasos viro-lógicos previos y pruebas genotípicas de resistencias disponibles, con el fin de tener en cuenta posibles mutaciones importantes archivadas, además de la existencia de comorbilidades (coinfección por virus de la hepatitis B [VHB], riesgo cardiovascular, función renal, densidad mineral ósea y otras coinfecciones) y su tratamiento. Es especialmente importante revisar la situación del paciente respecto al VHB: serología y situación vacunal10,16.
En general, se recomienda que el nuevo régimen incluya fármacos recomendados en las guías para pacientes que no han recibido tratamiento antirretroviral previamente, ya sea como preferentes o como «otras pautas antirretrovirales» en función del paciente. Es importante realizar una evaluación minuciosa del perfil de toxicidades, interacciones, restricciones dietéticas y actividad sobre el VHB, si fuera necesario, de la nueva pauta. En cuanto al número de fármacos, se puede mantener una pauta con 3 fármacos, estrategia con mayor experiencia y evidencia, sobre todo a raíz de la aparición de los distintos tratamientos en un solo comprimido con TAF o abacavir, o pasar a 2, con lamivudina o libres de análogos, cuya evidencia se está generando en estos momentos. No está recomendado pasar a un solo fármaco10,16-18.
Si la pauta contiene 3 fármacos, se puede realizar un cambio entre antirretrovirales de la misma clase (de efavirenz a rilpivirina o de raltegravir a dolutegravir [DTG] o elvitegravir [EVG], o de IP potenciado a tratamiento en comprimido único con DRV/c, o de TDF o abaca-vir a TAF, o de TDF a abacavir) o bien se puede realizar el cambio a otra clase (de IP potenciado con ritonavir a rilpivirina o a EVG, DTG; de inhibidores no nucleosídicos de la transcriptasa inversa [NNRTI, non-nucleoside reverse transcriptase inhibitor] a EVG o DTG). Hay que tener en cuenta la barrera genética de la nueva pauta, sobre todo si se cambia a fármacos de distinta clase, ya que hay que garantizar la actividad antirretroviral del TAR, tanto de los inhibidores nucleosídi-cos de la transcriptasa inversa (NRTI) como del tercer fármaco10.
Se asume que las personas sin fracaso virológico previo y sin mutaciones de resistencia con la carga viral plasmática suprimida no han generado nuevas mutaciones. El problema radica en aquellas que han presentado un fallo virológico previo. En ese caso se debe diseñar una nueva pauta cuya barrera genética sea similar a la de la previa. Esto es especialmente importante cuando se cambia a un TAR que incluye un IP potenciado; en esta situación, el especialista clínico debe diseñar una pauta que tenga en cuenta una posible resistencia archivada, bien sea confirmada o sospechada10,18.
Monitorización tras el cambioTras el cambio, el especialista clínico debe evaluar, en un plazo de 3-6 semanas, el mantenimiento de la supresión virológica y las determinaciones de laboratorio pertinentes que dependerán del motivo del cambio. Una vez que se ha demostrado la continuación de la supresión virológica, el paciente puede volver a la rutina de visitas cada 4 o 6 meses10,16,18.
Estudio EMERALDLa combinación de darunavir/cobicistat junto con 2 NRTI está incluida en las distintas guías de tratamiento como de elección, recomendada o alternativa en los pacientes que no han recibido tratamiento antirretroviral previamente, y como uno de los elementos fundamentales de las terapias de rescate10,16,17. DRV es un IP con barrera genética alta que puede proporcionar una respuesta virológica duradera sin aparición de mutaciones de resistencia en muchos pa-cientes19. Se comercializó hace unos 10 años, con lo que su experiencia en ensayos clínicos y práctica clínica diaria es amplia, y en la actualidad se ha mantenido como el IP de elección10,16-18. La eficacia y seguridad de la combinación de DRV/c y FTC/TAF en un solo comprimido en una estrategia de cambio se ha explorado en el estudio EMERALD (TMC114FD3013; NCT02269917)15.
Es un ensayo clínico15 de fase III, de distribución aleatoria (2:1), activo-control, abierto, internacional, multicéntrico, de no inferioridad que incluye a pacientes con VIH adultos, mayores de 18 años, pretratados con carga viral indetectable y un tratamiento estable con una combinación de FTC/TDF + IP potenciado, al menos, durante 6 meses, en que el IP potenciado es: DRV una vez al día, atazanavir o lopinavir. Se realizó en 106 centros en 9 países europeos y de Norteamérica.
Se definía a paciente suprimido como aquel que presentaba carga viral inferior a 50 copias/ml, al menos, los 2 últimos meses. Se permitía una carga viral de 50-200 copias/ml en los 12 meses previos al cribado15.
A diferencia de otros estudios de cambio de tratamiento, se permitía la inclusión de pacientes que hubiesen experimentado un fracaso virológico con pautas de tratamiento sin DRV y de aquellos con prueba genotípica previa en que se objetivaban mutaciones de resistencia, siempre que no fueran a DRV15.
Los pacientes se distribuyeron aleatoriamente 2:1 entre recibir la nueva combinación y seguir la pauta previa (grupo control), y se estratificaron en función del IP basal. El período de cribado fue alrededor de 30 días (6 semanas como máximo) y el período de estudio fue 48 semanas, tras lo cual se podía cambiar al STR en una fase de extensión. Las visitas se programaron tras la basal en las semanas 2, 4, 8 y 12, y cada 12 semanas15.
Como ya se ha mencionado, se trata de un estudio de no inferioridad, en el cual el objetivo primario fue la proporción de pacientes con rebrote virológico acumulado en la semana 48 con un margen del 4% de diferencia entre el tratamiento en estudio y el control, siguiendo los nuevos criterios de la FDA para los ensayos de cambio de tratamiento ya que los pacientes presentan carga viral indetecta-ble en el momento de la inclusión. Se entiende como rebrote virológico la presencia de una carga viral igual o superior a 50 copias/ml confirmada o las suspensiones prematuras, independientemente de la razón, con la última carga viral disponible igual o superior a 50 copias/ml15.
Los objetivos secundarios y exploratorios incluyeron seguridad y tolerabilidad, valoración de la actividad antirretroviral con el análisis snapshot de la FDA en la semana 48 (proporción de pacientes con cargas virales inferiores a 20, 50 y 200 copias/ml), tiempo hasta el rebrote virológico, cambio de CD4 desde la basal, aparición de resistencias genotípicas durante el ensayo clínico, adherencia y cambios en la creatinina sérica, filtrado glomerular y en los biomarcadores de función renal. Además, se puso en marcha un subestudio óseo, en el cual los objetivos fueron cambios en la DMO y en los biomarcadores óseos desde la basal.
El análisis primario realizado fue por intención de tratar (ITT) e incluyó a cualquier paciente que recibió, al menos, una dosis de la medicación.
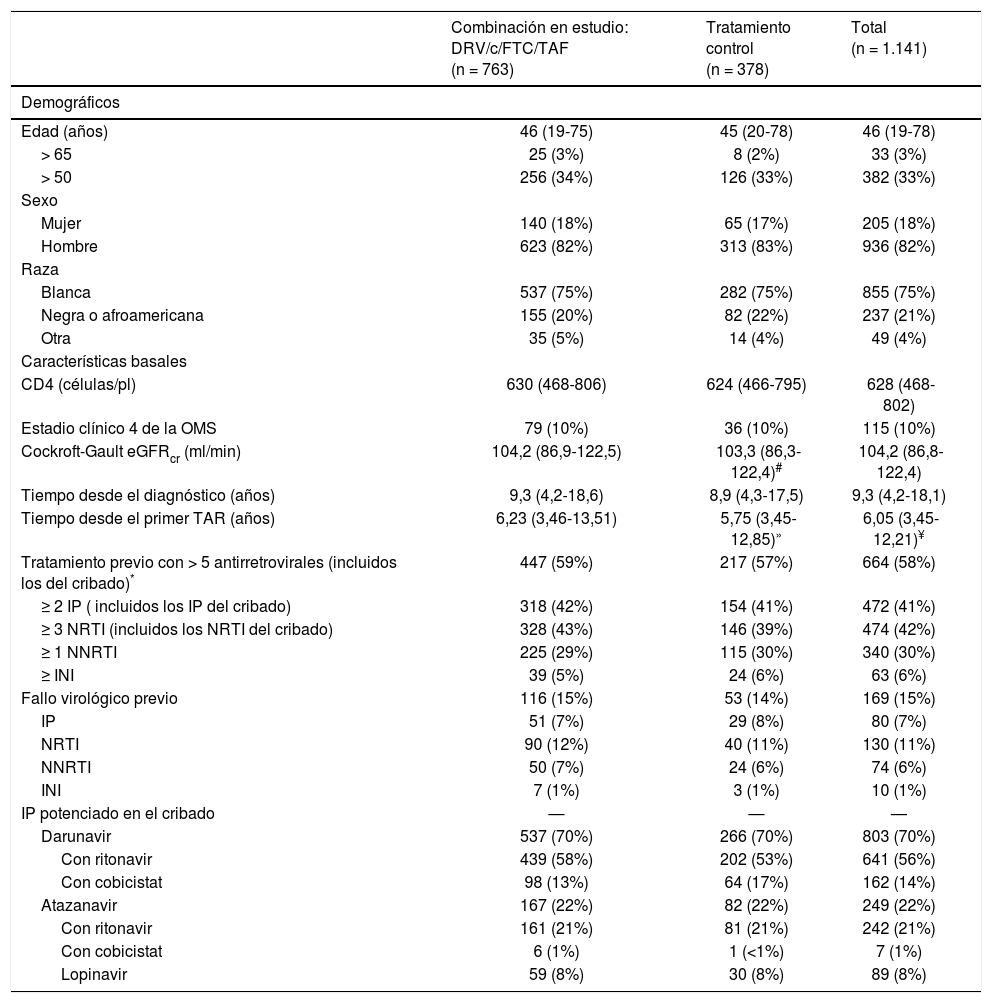
Características de los pacientesSe realizó el cribado a 1.299 pacientes, de los cuales se distribuyó aleatoriamente a 1.141: 763 en el grupo de estudio y 378 en el control15. Los 2 grupos presentaban unas características basales bien equilibradas: la mayoría eran varones (82%; n = 936) y de raza blanca (75%; n = 855), con una media de edad de 46 años; el 33% (n = 382) eran mayores de 50 añ15.
En cuanto al tratamiento antirretroviral previo, el 58% (n = 668/1.141) habían sido pretratados, al menos, con una línea de tratamiento antes del cribado; el 15% (n = 169) habían tenido fracaso virológico previo no a DRV y el 70% (n = 803) estaban tomando DRV potenciado durante el cribado. Además, el 58% (n = 664) habían recibido ya, al menos, 5 an-tirretrovirales y el 41% (n = 472), al menos, 2 IP (tabla 1).
Características basales de los pacientes: aspectos demográficos y relacionados con el VIH
| Combinación en estudio: DRV/c/FTC/TAF (n = 763) | Tratamiento control (n = 378) | Total (n = 1.141) | |
|---|---|---|---|
| Demográficos | |||
| Edad (años) | 46 (19-75) | 45 (20-78) | 46 (19-78) |
| > 65 | 25 (3%) | 8 (2%) | 33 (3%) |
| > 50 | 256 (34%) | 126 (33%) | 382 (33%) |
| Sexo | |||
| Mujer | 140 (18%) | 65 (17%) | 205 (18%) |
| Hombre | 623 (82%) | 313 (83%) | 936 (82%) |
| Raza | |||
| Blanca | 537 (75%) | 282 (75%) | 855 (75%) |
| Negra o afroamericana | 155 (20%) | 82 (22%) | 237 (21%) |
| Otra | 35 (5%) | 14 (4%) | 49 (4%) |
| Características basales | |||
| CD4 (células/pl) | 630 (468-806) | 624 (466-795) | 628 (468-802) |
| Estadio clínico 4 de la OMS | 79 (10%) | 36 (10%) | 115 (10%) |
| Cockroft-Gault eGFRcr (ml/min) | 104,2 (86,9-122,5) | 103,3 (86,3-122,4)# | 104,2 (86,8-122,4) |
| Tiempo desde el diagnóstico (años) | 9,3 (4,2-18,6) | 8,9 (4,3-17,5) | 9,3 (4,2-18,1) |
| Tiempo desde el primer TAR (años) | 6,23 (3,46-13,51) | 5,75 (3,45-12,85)» | 6,05 (3,45-12,21)¥ |
| Tratamiento previo con > 5 antirretrovirales (incluidos los del cribado)* | 447 (59%) | 217 (57%) | 664 (58%) |
| ≥ 2 IP ( incluidos los IP del cribado) | 318 (42%) | 154 (41%) | 472 (41%) |
| ≥ 3 NRTI (incluidos los NRTI del cribado) | 328 (43%) | 146 (39%) | 474 (42%) |
| ≥ 1 NNRTI | 225 (29%) | 115 (30%) | 340 (30%) |
| ≥ INI | 39 (5%) | 24 (6%) | 63 (6%) |
| Fallo virológico previo | 116 (15%) | 53 (14%) | 169 (15%) |
| IP | 51 (7%) | 29 (8%) | 80 (7%) |
| NRTI | 90 (12%) | 40 (11%) | 130 (11%) |
| NNRTI | 50 (7%) | 24 (6%) | 74 (6%) |
| INI | 7 (1%) | 3 (1%) | 10 (1%) |
| IP potenciado en el cribado | — | — | — |
| Darunavir | 537 (70%) | 266 (70%) | 803 (70%) |
| Con ritonavir | 439 (58%) | 202 (53%) | 641 (56%) |
| Con cobicistat | 98 (13%) | 64 (17%) | 162 (14%) |
| Atazanavir | 167 (22%) | 82 (22%) | 249 (22%) |
| Con ritonavir | 161 (21%) | 81 (21%) | 242 (21%) |
| Con cobicistat | 6 (1%) | 1 (<1%) | 7 (1%) |
| Lopinavir | 59 (8%) | 30 (8%) | 89 (8%) |
Si revisamos las comorbilidades, el 7% de los pacientes (n = 82/1.141) presentaban historia previa de diabetes/hiperglucemia y el 23% (n = 262) presentaban historia previa de hipertensión20.
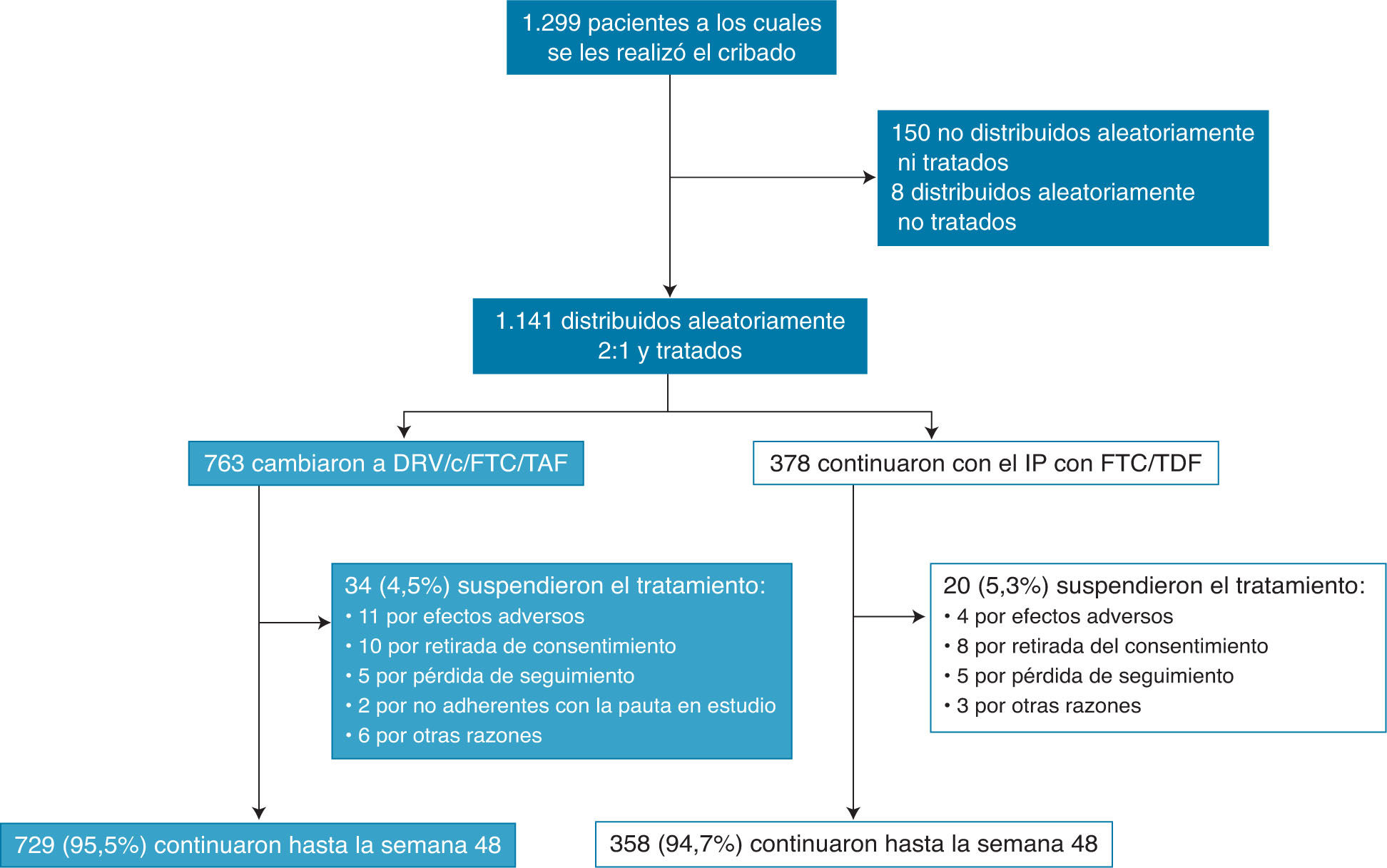
Se publicaron los datos a 48 semanas y se incluyó a 1.087 (95%) de los 1.141 en el análisis por intención de tratar (fig. 1)15.
: a phase 3, randomised, non-inferiority trial”, Pages e23-e34, © Elsevier Ltd. (2017), con permiso de Elsevier. DRV/c/FTC/TAF, darunavir, cobicistat, emtricitabina y tenofovir alafenamida; FTC/TDF, emtricitabina y tenofovir disoproxil; IP, inhibidor de la proteasa.")
Diagrama que recoge la evolución de la participación de los pacientes incluidos en el estudio. Reproducida de The Lancet, Vol. 5, Núm. 1, C. Orkin, J.M. Molina, E. Negre-do, J.R. Arribas, J. Gathe, J.J. Eron, et al. “Efficacy and safety of switching from boosted protease inhibitors plus emtricitabine and tenofovir disoproxil fumarate regimens to single-tablet darunavir, cobicistat, emtricitabine, and tenofovir alafenamide at 48 weeks in adults with virologically suppressed HIV-1 (EMERALD): a phase 3, randomised, non-inferiority trial”, Pages e23-e34, © Elsevier Ltd. (2017), con permiso de Elsevier.
DRV/c/FTC/TAF, darunavir, cobicistat, emtricitabina y tenofovir alafenamida; FTC/TDF, emtricitabina y tenofovir disoproxil; IP, inhibidor de la proteasa.
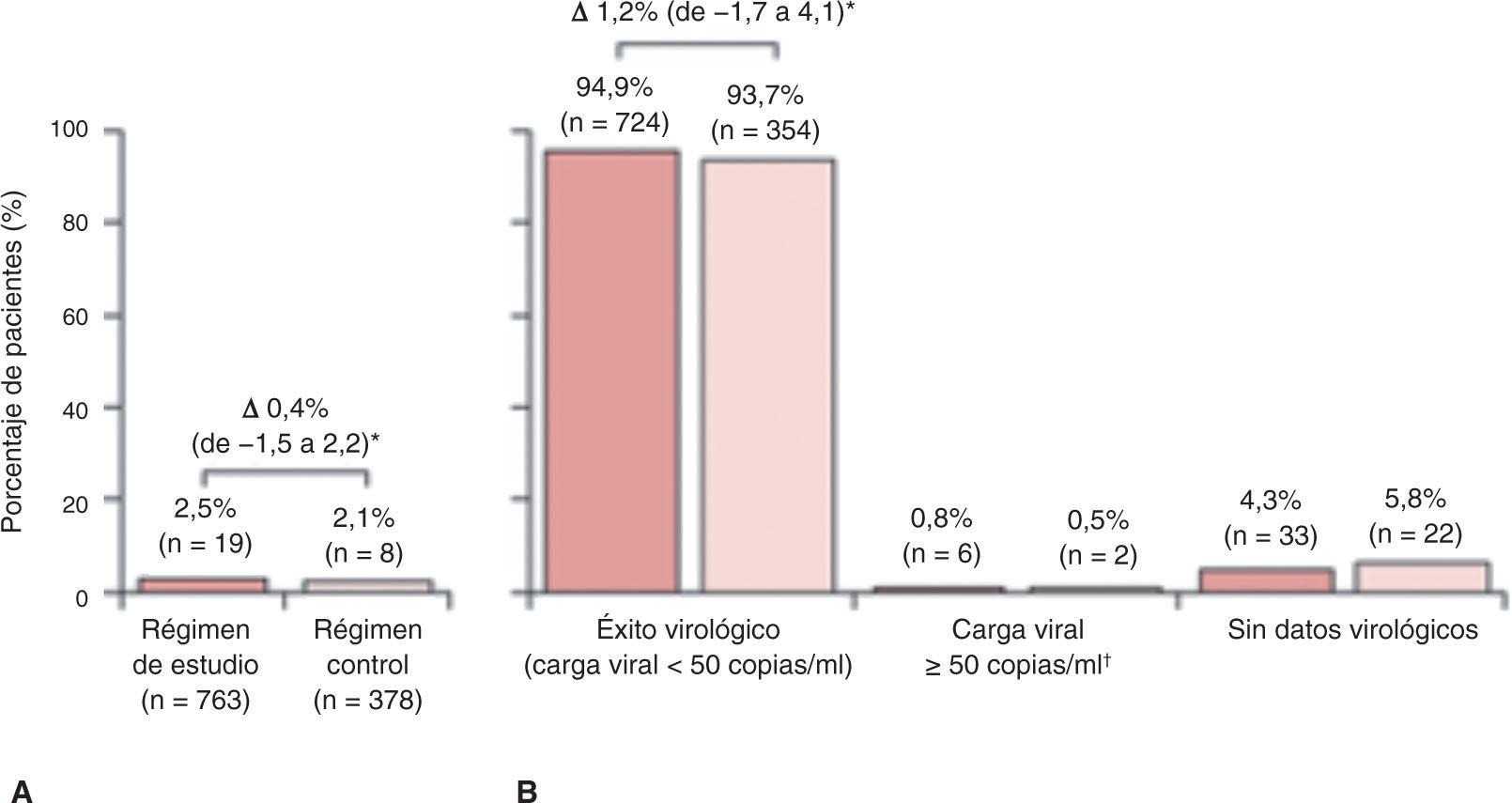
La proporción de pacientes con rebrote virológico confirmado en la semana 4815,21,22 fue baja y similar en ambos grupos: el 2,5% (19/763) en la pauta en estudio y el 2,1% (8/378) en el grupo control, con una diferencia del 0,4% (intervalo de confianza al 95% [IC95%] del -1,5 al 2,2%; p< 0,0001). Se demuestra la no inferioridad de la combinación en estudio frente al grupo control al ser la diferencia menor al 4% según los criterios de la FDA. De aquellos que experimentaron rebrote virológico, el 63% (12 de 19) del grupo de estudio y el 50% (4 de 8) del grupo control alcanzaron la respuesta virológica en la semana 48 sin ajuste de tratamiento. Tres pacientes del régimen en estudio experimentaron rebrotes confirmados ≥ 200 copias/ml, comparado con ninguno en la rama control. El análisis por protocolo confirma la no inferioridad (fig. 2)15,21,22.
 y análisis por snapshot en la semana 48 (B). Reproducida de The Lancet, Vol. 5, Núm. 1, C. Orkin, J.M. Molina, E. Negredo, J.R. Arribas, J. Gathe, J.J. Eron, et al. “Efficacy and safety of switching from boosted protease inhibitors plus emtricitabine and tenofovir disoproxil fumarate regimens to single-tablet darunavir, cobicistat, emtricitabine, and tenofovir alafenamide at 48 weeks in adults with virologically suppressed HIV-1 (EMERALD): a phase 3, randomised, non-inferiority trial”, Pages e23-e34, © Elsevier Ltd. (2017), con permiso de Elsevier. El régimen en estudio es darunavir, cobicistat, emtricitabina y tenofovir alafenamida (DRV/c/FTC/TAF). El régimen control es un inhibidor de la proteasa (IP) potenciado con te-nofovir disoproxil y emtricitabina (TDF/FTC). *Se calculó la diferencia de proporciones con un intervalo de confianza al 95% con la prueba de Mantel-Haenzel, con ajuste por el IP basal. †Última carga viral en la ventana de la semana 48 igual o superior a 50 copias/ml, o suspensiones por razones de eficacia o suspensiones prematuras no debidas a eficacia, efectos adversos o muerte con una única carga viral igual o superior a 50 copias/ml.")
Rebrote virológico confirmado acumulado hasta la semana 48 (A) y análisis por snapshot en la semana 48 (B). Reproducida de The Lancet, Vol. 5, Núm. 1, C. Orkin, J.M. Molina, E. Negredo, J.R. Arribas, J. Gathe, J.J. Eron, et al. “Efficacy and safety of switching from boosted protease inhibitors plus emtricitabine and tenofovir disoproxil fumarate regimens to single-tablet darunavir, cobicistat, emtricitabine, and tenofovir alafenamide at 48 weeks in adults with virologically suppressed HIV-1 (EMERALD): a phase 3, randomised, non-inferiority trial”, Pages e23-e34, © Elsevier Ltd. (2017), con permiso de Elsevier.
El régimen en estudio es darunavir, cobicistat, emtricitabina y tenofovir alafenamida (DRV/c/FTC/TAF). El régimen control es un inhibidor de la proteasa (IP) potenciado con te-nofovir disoproxil y emtricitabina (TDF/FTC).
*Se calculó la diferencia de proporciones con un intervalo de confianza al 95% con la prueba de Mantel-Haenzel, con ajuste por el IP basal. †Última carga viral en la ventana de la semana 48 igual o superior a 50 copias/ml, o suspensiones por razones de eficacia o suspensiones prematuras no debidas a eficacia, efectos adversos o muerte con una única carga viral igual o superior a 50 copias/ml.
El tiempo hasta la aparición del rebrote fue similar en los 2 grupos y estuvo repartido de forma homogénea a lo largo de las 48 semanas15.
El análisis por snapshot de la FDA demuestra también tasas muy similares de eficacia virológica entre el tratamiento en estudio (94,9%) y el control (93,7%), con una diferencia de 1,2% (IC95%: del-1,7 al 4,1%) que favorece al régimen en estudio15,21,22. Solo 6 pacientes (0,8%) de la rama del estudio y 2 (0,5) de la rama control tenían una carga viral superior a 50 copias/ml en la semana 48 (diferencia del 0,3%; IC95%: del -0,7 al 1,2%)15,21,22.
En el análisis por snapshot por protocolo, el 96,3% (694/721) de los pacientes del grupo de estudio y el 95,5% (342/358) de los pacientes del grupo control presentaban una carga viral inferior a 50 copias/ml15,21,22.
Se han realizado distintos análisis por subgrupos basales: edad, sexo, raza, existencia de fracasos virológicos previos y por experiencia previa a antirretrovirales, e incluso según el IP basal y el potenciador; en todos ellos se reproducen los mismos resultados de no inferioridad y de elevada eficacia virológica de DRV/c/FTC/TAF23-26.
En cuanto al incremento de CD4 fue similar en los 2 grupos y no se objetivaron diferencias importantes15.
ResistenciasMuy pocos pacientes presentaron rebrote virológico y la mayoría de ellos tenían cargas virales bajas (< 400 copias/ml), por lo que solo se pudieron genotipar muestras de un paciente de la rama de Symtu-za® y 3 de la rama control. En ninguno de ellos se detectaron mutaciones primarias de IP ni mutaciones de resistencia a darunavir, em-tricitabina ni tenofovir15.
AdherenciaAl final de la semana 48, la adherencia acumulada medida por recuento de tabletas fue el 99,7% (amplitud intercuartílica [IQR]: 98,5-100,3) en el grupo de estudio y el 99,3% (97,4-100) en el grupo control15.
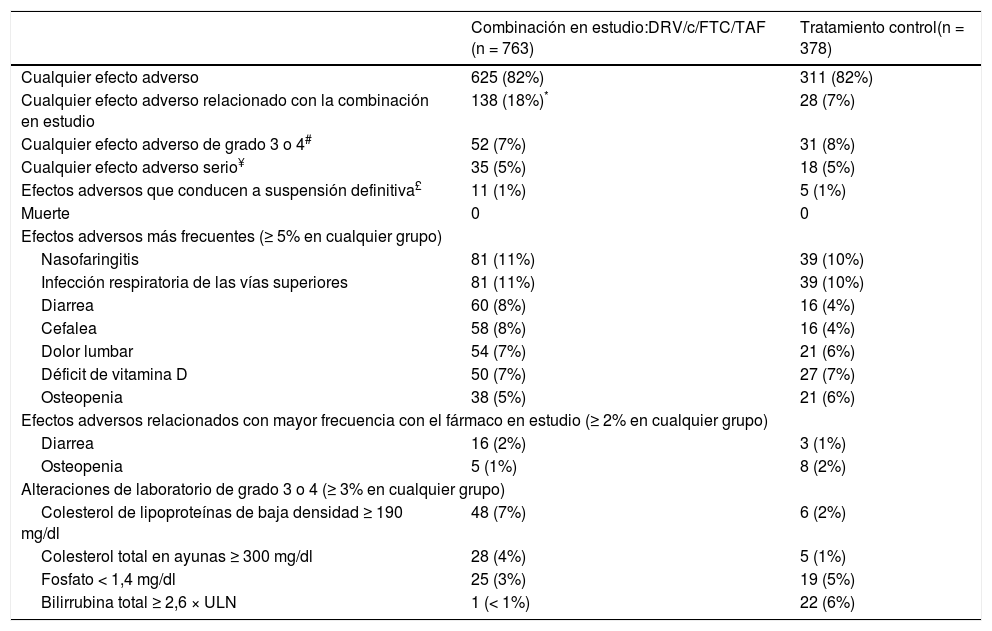
SeguridadFue similar en ambos grupos. Se observó mayor incidencia de efectos adversos (AES) relacionados con el fármaco en el grupo de estudio (el 18 frente al 7%). La mayoría fue grado 1-2, muy pocos grados 3 y 4, efectos adversos serios (SAES) o suspensiones por efectos adversos15. Las tasas de AES y SAES fueron similares en los 2 grupos (82 y 5%, respectivamente). La frecuencia de la mayoría de AES fue similar en los 2 grupos, con excepción de los cambios medios en las cifras de coles-terol de grado 3-4 que fueron mayores en la rama de estudio. Se observó una pequeña diferencia, pero estadísticamente significativa, en el cambio medio desde la basal en el grupo de estudio frente al 0,1% (control); p = 0,01. De hecho, durante el ensayo clínico se inició tratamiento hipolipemiante en 20 (3%) de 763 pacientes en la rama de estudio frente a 7 (2%) de 378 de la rama control (p = 0,54) (tabla 2).
Efectos adversos y alteraciones de resultados de laboratorio en la semana 48
| Combinación en estudio:DRV/c/FTC/TAF (n = 763) | Tratamiento control(n = 378) | |
|---|---|---|
| Cualquier efecto adverso | 625 (82%) | 311 (82%) |
| Cualquier efecto adverso relacionado con la combinación en estudio | 138 (18%)* | 28 (7%) |
| Cualquier efecto adverso de grado 3 o 4# | 52 (7%) | 31 (8%) |
| Cualquier efecto adverso serio¥ | 35 (5%) | 18 (5%) |
| Efectos adversos que conducen a suspensión definitiva£ | 11 (1%) | 5 (1%) |
| Muerte | 0 | 0 |
| Efectos adversos más frecuentes (≥ 5% en cualquier grupo) | ||
| Nasofaringitis | 81 (11%) | 39 (10%) |
| Infección respiratoria de las vías superiores | 81 (11%) | 39 (10%) |
| Diarrea | 60 (8%) | 16 (4%) |
| Cefalea | 58 (8%) | 16 (4%) |
| Dolor lumbar | 54 (7%) | 21 (6%) |
| Déficit de vitamina D | 50 (7%) | 27 (7%) |
| Osteopenia | 38 (5%) | 21 (6%) |
| Efectos adversos relacionados con mayor frecuencia con el fármaco en estudio (≥ 2% en cualquier grupo) | ||
| Diarrea | 16 (2%) | 3 (1%) |
| Osteopenia | 5 (1%) | 8 (2%) |
| Alteraciones de laboratorio de grado 3 o 4 (≥ 3% en cualquier grupo) | ||
| Colesterol de lipoproteínas de baja densidad ≥ 190 mg/dl | 48 (7%) | 6 (2%) |
| Colesterol total en ayunas ≥ 300 mg/dl | 28 (4%) | 5 (1%) |
| Fosfato < 1,4 mg/dl | 25 (3%) | 19 (5%) |
| Bilirrubina total ≥ 2,6 × ULN | 1 (< 1%) | 22 (6%) |
Definido por el sistema de gradación de los efectos adversos de la división de Sida, estandarizado.
Solo se describió un efecto adverso serio relacionado posiblemente con la combinación en estudio por el investigador: una pancreatitits aguda.
Se identificaron efectos adversos graves relacionados con la combinación en estudio según el investigador en 8 pacientes (1%; incluyendo 3 episodios gastrointestinales, una cefalea, un episodio psiquiátrico, un incremento de tran-saminasas, un caso de urticaria y un episodio renal), y en 3 pacientes de la rama de control (1%; uno con episodios gastrointestinales, astenia, visión borrosa y en túnel, y 2 pacientes con episodios renales). ULN, nivel superior de la normalidad.
En cuanto a los efectos adversos renales, ocurrieron en 30 (4%) de 763 participantes que tomaban STR frente a 18 (5%) de 378 en la rama control. Tres episodios condujeron a la suspensión, uno en el grupo de estudio (empeoramiento de una insuficiencia renal crónica preexistente de grado 2) y 2 en el control.
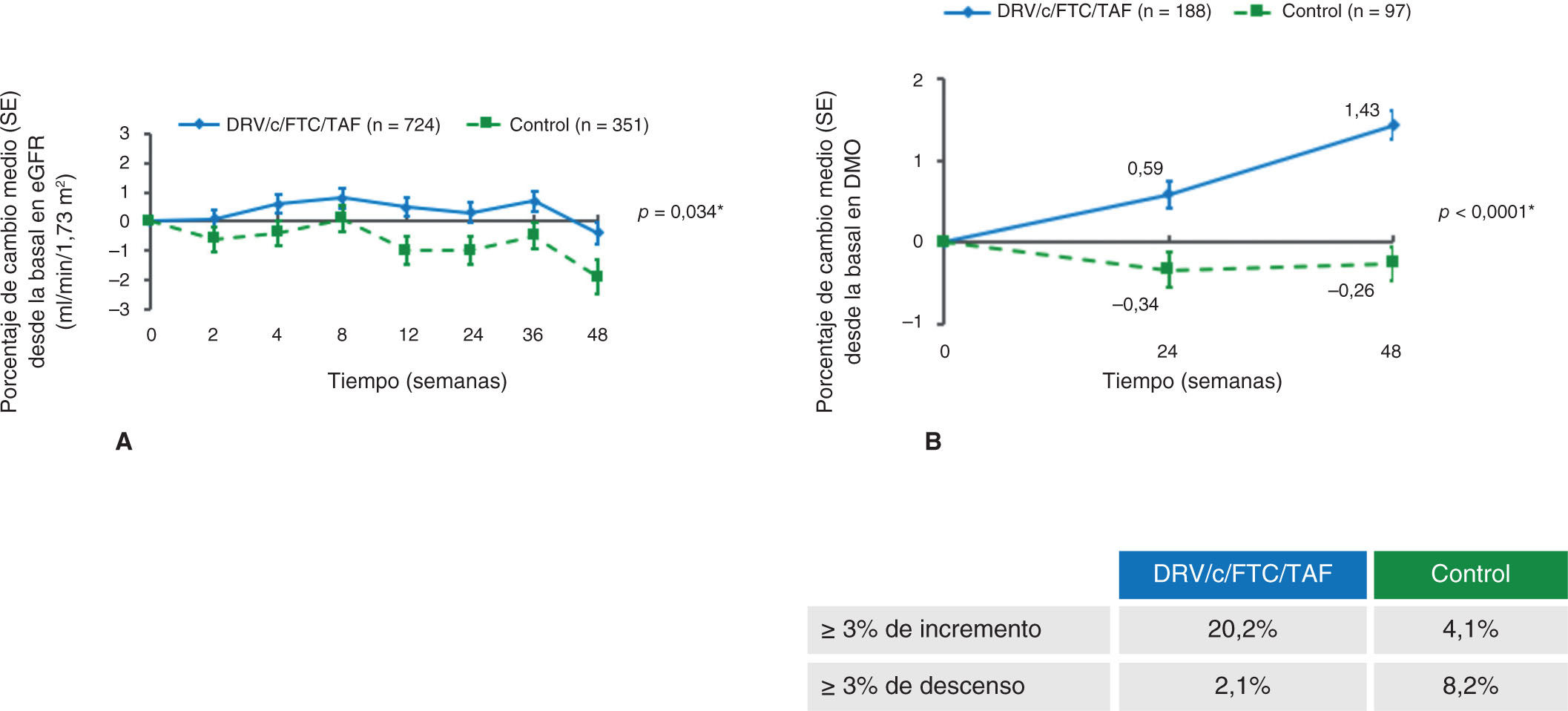
En cuanto a la función renal, se observó una disminución del filtrado glomerular basado en la cistatina sérica en el grupo control mientras que permanecía estable en el grupo con TAF, diferencia que fue importante en la semana 48 (p = 0,034)15 (fig. 3 A). No se observan diferencias considerables cuando lo que se analiza es la creatini-na sérica y el filtrado glomerular estimado15.
 Filtrado glomerular estimado basado en cistatina C. *Valor p para la diferencia estimado usando ANCOVA, incluyendo el tratamiento como factor y el filtrado glomerular basal como un covariado. eGFR, receptor del factor de crecimiento epidérmico. B) Porcentaje medio de cambios en la densidad mineral ósea (DMO) en la semana 48. Cadera. *Valor p para ANCOVA incluyendo densidad mineral ósea basal e inhibidor de la proteasa potenciado en el cribado como covariado. Reproducida de The Lancet, Vol. 5, Núm. 1, C. Orkin, J.M. Molina, E. Negredo, J.R. Arribas, J. Gathe, J.J. Eron, et al. “Efficacy and safety of switching from boosted protease inhibitors plus emtricitabine and tenofovir disoproxil fumarate regimens to single-tablet darunavir, cobicistat, emtricitabine, and tenofovir alafenamide at 48 weeks in adults with virologically suppressed HIV-1 (EMERALD): a phase 3, randomised, non-inferiority trial”, Pages e23-e34, © Elsevier Ltd. (2017), con permiso de Elsevier.")
Cambio medio desde la basal a la semana 48 en los parámetros renales y óseos. A) Filtrado glomerular estimado basado en cistatina C. *Valor p para la diferencia estimado usando ANCOVA, incluyendo el tratamiento como factor y el filtrado glomerular basal como un covariado. eGFR, receptor del factor de crecimiento epidérmico. B) Porcentaje medio de cambios en la densidad mineral ósea (DMO) en la semana 48. Cadera. *Valor p para ANCOVA incluyendo densidad mineral ósea basal e inhibidor de la proteasa potenciado en el cribado como covariado. Reproducida de The Lancet, Vol. 5, Núm. 1, C. Orkin, J.M. Molina, E. Negredo, J.R. Arribas, J. Gathe, J.J. Eron, et al. “Efficacy and safety of switching from boosted protease inhibitors plus emtricitabine and tenofovir disoproxil fumarate regimens to single-tablet darunavir, cobicistat, emtricitabine, and tenofovir alafenamide at 48 weeks in adults with virologically suppressed HIV-1 (EMERALD): a phase 3, randomised, non-inferiority trial”, Pages e23-e34, © Elsevier Ltd. (2017), con permiso de Elsevier.
La densidad mineral ósea (DMO) en cadera y columna también disminuyó en el grupo control, mientras que aumentó en el de estudio, con diferencias considerables en ambas localizaciones en la semana 48 (p < 0,0001) (figura 3B).
Solo un SAE en el grupo de estudio se comunicó como posiblemente relacionado con el fármaco15.
Subanálisis de hueso y riñónPosteriormente se realizó un análisis de subgrupos post hoc de este ensayo para estudiar mejor los efectos adversos a nivel renal y óseo tras cambiar a DRV/c/FCT/TAF20. Se diseñaron 2 subestudios, óseo y renal, cuyos objetivos fueron, respectivamente, evaluar el impacto de la edad (> 50 y ≤ 50 años) y el sexo sobre la DMO, y el impacto de la edad, la existencia previa de diabetes mellitus/hiperglucemia e hipertensión sobre la función renal y la proteinuria.
En el subestudio óseo se incluyó a 209 pacientes con STR frente a 108 controles, con las características basales similares a las del estudio global, a los cuales se les realizó radioabsorciometría de energía dual (DEXA) a la altura de la columna lumbar (L1-L4) y de cadera, en el momento basal, semana 24 y semana 48. Se calculó el porcentaje de cambio en las semanas 24 y 48.
En el subestudio renal, los efectos adversos renales fueron moni-torizados y la función renal determinada en cada visita. Se calcularon los cambios desde el basal hasta la semana 48 en el receptor del factor de crecimiento epidérmico (eGFR) basado en la cistatina sérica (eGFRcyst; fórmula CKD-EPI) y los índices de proteinuria total, proteína fijadora de retinol (en ayunas) y β2-microglobulina (en ayunas)/creatinina (UPCR, RBP:Cr y β2-microglobulina/creatinina).
La DMO de la columna y la cadera de los pacientes que cambiaron a TAF mejoró y empeoró en los que mantuvieron el tratamiento, independientemente de la edad y el sexo. Una tendencia similar se observó en los cambios en el cuello femoral.
En cuanto al estudio renal, en todos los subgrupos los cambios observados en eGFRcys desde la basal hasta la semana 48 en la rama de TAF no fueron estadísticamente significativos y el efecto del cambio a STR sobre los cambios en eGFRcys fueron coherentes. Comparado con TAF, en general, los controles presentaban diferencias modestas, pero estadísticamente significativas, en los mayores de 50 años o sin diabetes mellitus o hipertensión previa.
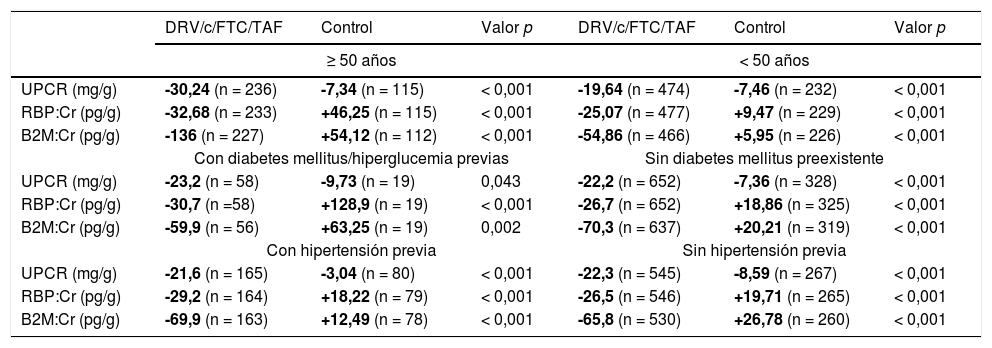
Si evaluamos la proteinuria, el cambio a TAF se tradujo en mejoras importantes en la semana 48 en todas las medidas de proteinuria cuantitativa, independientemente de la edad, diabetes/hipergluce-mia e hipertensión arterial existentes previamente en todos los sub-grupos (tabla 3).
Cambios medios en la proteinuria en la semana 48
| DRV/c/FTC/TAF | Control | Valor p | DRV/c/FTC/TAF | Control | Valor p | |
|---|---|---|---|---|---|---|
| ≥ 50 años | < 50 años | |||||
| UPCR (mg/g) | -30,24 (n = 236) | -7,34 (n = 115) | < 0,001 | -19,64 (n = 474) | -7,46 (n = 232) | < 0,001 |
| RBP:Cr (pg/g) | -32,68 (n = 233) | +46,25 (n = 115) | < 0,001 | -25,07 (n = 477) | +9,47 (n = 229) | < 0,001 |
| B2M:Cr (pg/g) | -136 (n = 227) | +54,12 (n = 112) | < 0,001 | -54,86 (n = 466) | +5,95 (n = 226) | < 0,001 |
| Con diabetes mellitus/hiperglucemia previas | Sin diabetes mellitus preexistente | |||||
| UPCR (mg/g) | -23,2 (n = 58) | -9,73 (n = 19) | 0,043 | -22,2 (n = 652) | -7,36 (n = 328) | < 0,001 |
| RBP:Cr (pg/g) | -30,7 (n =58) | +128,9 (n = 19) | < 0,001 | -26,7 (n = 652) | +18,86 (n = 325) | < 0,001 |
| B2M:Cr (pg/g) | -59,9 (n = 56) | +63,25 (n = 19) | 0,002 | -70,3 (n = 637) | +20,21 (n = 319) | < 0,001 |
| Con hipertensión previa | Sin hipertensión previa | |||||
| UPCR (mg/g) | -21,6 (n = 165) | -3,04 (n = 80) | < 0,001 | -22,3 (n = 545) | -8,59 (n = 267) | < 0,001 |
| RBP:Cr (pg/g) | -29,2 (n = 164) | +18,22 (n = 79) | < 0,001 | -26,5 (n = 546) | +19,71 (n = 265) | < 0,001 |
| B2M:Cr (pg/g) | -69,9 (n = 163) | +12,49 (n = 78) | < 0,001 | -65,8 (n = 530) | +26,78 (n = 260) | < 0,001 |
Mientras que los cambios en el control fueron mayores que en el grupo de estudio para algunos subgrupos, los resultados se mantuvieron en los límites normales.
La conclusión de estos subestudios es que el cambio a STR mejora los parámetros de seguridad ósea y los biomarcadores de proteinuria independientemente de la edad, el sexo y la preexistencia de diabe-tes/hiperglucemia y de hipertensión arterial, que son los factores que pueden aumentar el riesgo de enfermedad renal y ósea en los pacientes con VIH20.
ConclusionesEl estudio EMERALD es un estudio de no inferioridad que demuestra la eficacia y seguridad del comprimido único de darunavir, cobicistat, emtricitabina y tenofovir alafenamida como opción de cambio de tratamiento para pacientes con VIH y carga viral suprimida15,20-26.
Este es el primer ensayo realizado con un STR que contiene un IP potenciado, en este caso, darunavir, y el primer ensayo de cambio en que se valora la respuesta con los criterios nuevos de la FDA, mucho más estrictos15.
Además, es un ensayo de cambio de tratamiento atípico ya que permite la inclusión de pacientes muy variados, incluso con fracasos previos a otros grupos de fármacos o mutaciones previas, siempre que no sean a DRV y experiencia previa con IP15.
En cuanto a los resultados, se demuestra la no inferioridad, con escaso número de rebrotes, eficacia virológica elevada y la no aparición de mutaciones de resistencia. La tolerancia en ambos brazos del estudio ha sido muy buena y se ha objetivado mejoría en la DMO y la proteinuria en la rama de TAF15,20-26.
La principal limitación del estudio es que se trata de un diseño abierto y que algunos grupos de pacientes están infrarrepresentados. La ventaja es que refleja mejor la población de pacientes reales que podrían ser candidatos a cambio frente a otros estudios, en los cuales no se admiten fracasos previos y son más restrictivos15.
¿Qué beneficios aporta Symtuza® en pacientes tratados previamente?Symtuza® es el primer STR que contiene TAF y un IP, en este caso, darunavir; es una combinación de antirretrovirales efectiva, bien tolerada y cómoda, que combina la potencia y la barrera genética elevada del DRV con las ventajas a nivel renal y óseo del TAF. En el ensayo EMERALD se pone de manifiesto que pacientes con carga viral indetectable y experiencia previa con antirretrovirales, en algunos casos muy amplia, e incluso con fracasos virológicos, pero sin mutaciones de resistencia a DRV, pueden permanecer indetectables de forma prolongada tras el cambio de tratamiento.
Conflicto de interesesLa autora, María José Galindo, ha actuado como consultora, ha recibido ayudas para investigación y ha participado en actividades científicas y proyectos educativos que han contado con el patrocinio de los laboratorios Bristol-Mayers Squibb, Abbvie, ViiV, Gilead, MSD, Jannsen-Cilag y Pfizer.
Información sobre el suplementoEste artículo forma parte del suplemento titulado «Darunavir, co-bicistat, emtricitabina y tenofovir alafenamida coformulados en el tratamiento de la infección por el VIH», que ha sido patrocinado por Janssen.

