





El abordaje de la infección por el VIH se basa en la administración del tratamiento antirretroviral (TAR) de forma indefinida. Los regímenes disponibles en un solo comprimido (STR, single tablet regimen) reducen al máximo la posología y facilitan la adherencia a largo plazo. La coformulación de darunavir/cobicistat/ emtricitabina/tenofovir alafenamida (DRV/c/FTC/TAF), con el nombre comercial de Symtuza®, es el primer STR basado en un inhibidor de la proteasa (IP). Symtuza® aporta la eficacia, potencia y alta barrera genética de DRV/c que lo posicionan como fármaco de elección en individuos con riesgo de desarrollar mutaciones de resistencia, junto con el buen perfil de seguridad de TAF y las ventajas de un STR. Permite, además, el inicio precoz del TAR al no tener que disponer del resultado de genotipo basal ni de HLA-B5701. Por ello es un régimen muy competitivo en pacientes que no han recibido TAR previamente, sobre todo con riesgo de mala adherencia y bajo potencial de interacciones farmacológicas. Información sobre el suplemento: este artículo forma parte del suplemento titulado «Darunavir, cobicistat, emtricitabina y tenofovir alafena-mida coformulados en el tratamiento de la infección por el VIH», que ha sido patrocinado por Janssen.
The management of HIV infection is based on the administration of lifelong antiretroviral therapy (ART). Single-tablet regimens (STR) reduce pill burden and maximise long-term adherence. Cobicistat-boosted darunavir with emtricitabine and tenofovir alafenamide co-formulation (DRV/c/FTC/TAF), with trade name Symtuza®, is the first STR based on a protease inhibitor (PI). Symtuza® exhibits the efficacy, potency and high genetic barrier of DRV/c, positioning it as the drug of choice even in patients at risk of developing resistance mutations, in addition to the good safety profile of TAF and the advantages of an STR. Early ART initiation is also possible as baseline genotype and HLA-B5701 are not needed. It therefore represents a very good regimen for naive patients, in particular those at risk of poor adherence, and those with low potential risk for drug-drug interactions. Supplement information: This article is part of a supplement entitled “Co-formulated cobicistat-boosted darunavir, emtricitabine, and tenofovir alafenamide for the treatment of HIV infection”, which is sponsored by Janssen.
Desde el inicio de la epidemia de la infección por el VIH, hace ya más de 3 décadas, se han producido grandes cambios tanto epidemiológicos como clínicos y terapéuticos. La introducción y generalización del tratamiento antirretroviral de gran actividad (TAR) con la llegada de los inhibidores de la proteasa (IP) a mediados de la década de los noventa del siglo pasado realmente cambiaron la historia natural de la enfermedad, con una drástica disminución de la mor-bimortalidad, y convirtieron la infección por el VIH en una enfermedad crónica1. Los IP de primera generación poseían una elevada eficacia virológica, pero mala tolerancia con efectos adversos que llevaban con frecuencia a la suspensión del TAR y con posologías de varias tomas al día con considerable número de pastillas2-5. Atazana-vir y darunavir (DRV), los IP de segunda generación, poseen un perfil de tolerancia mucho mejor con posologías de una sola toma al día6,7. La eficacia y seguridad a largo plazo, así como su elevada barrera genética, han posicionado a DRV como el IP de elección8-11. La coformulación de DRV con cobicistat, emtricitabina y tenofovir ala-fenamida (DRV/c/FTC/TAF) es el primer régimen basado en IP disponible en un solo comprimido (STR) y lo hace competitivo con otras pautas en STR.
Objetivos ante un paciente que no ha recibido tratamiento antirretroviral previamenteActualmente se recomienda TAR a todos los pacientes con infección por el VIH, si bien se individualiza el momento de inicio de este, que debe ser lo más precoz posible, así como el régimen que debe utilizarse10,11. Los objetivos principales del TAR son la preservación y recuperación inmunológicas, y el control de la replicación del virus, con lo que se consigue una reducción de la morbimortalidad asociadas con la infección por el VIH, se evita el efecto nocivo de la replica-ción viral sobre otras comorbilidades y se previene la transmisión del virus. En el momento de pautar un TAR de inicio, además de la eficacia, se tienen en cuenta otras premisas. Por un lado, que posea un buen perfil de seguridad y tolerancia, con pocos efectos adversos tanto clínicos como analíticos y que sean bien conocidos y manejables. Por otro lado, es muy importante la posología ya que está demostrado que las pautas más sencillas se asocian con mejor adherencia y, en este sentido, la máxima comodidad en posología la aportan los regímenes disponibles en STR12,13. La durabilidad, que en definitiva va a estar condicionada por la eficacia y la seguridad, y el bajo potencial de interacciones son otras 2 características que hay que tener presentes a la hora de iniciar el TAR. Las pautas recomendadas por todas las guías vigentes se basan en 2 inhibidores nucleosídicos de la transcriptasa inversa (NRTI, nucleoside reverse transcriptase inhibitor) combinados con un inhibidor de la integrasa (InI), con un IP potenciado o con un inhibidor no nucleosídico de la transcriptasa inversa (NNRTI, non-nucleoside reverse transcriptase inhibitor)10,11; con cualquiera de ellas, más del 80% de los pacientes alcanzan la supresión virológica a las 48 semanas. Las particularidades y requisitos de cada pauta junto con las características y circunstancias de cada paciente van a condicionar la elección de un régimen u otro de TAR de inicio.
¿Por qué un IP hoy en pacientes que no han recibido tratamiento antirretroviral previamente?Aunque podríamos afirmar que nos encontramos en la era de los InI y que las pautas basadas en este grupo son las actualmente recomendadas como preferentes para inicio del TAR10,11, hay bastantes pacientes que se beneficiarían de las características de los IP, concretamente de DRV, que es el IP de elección hoy día10,11. En primer lugar, es un fármaco con el cual se tiene una dilatada experiencia en la práctica clínica. Su potencia y elevada barrera genética dan tal robustez al TAR que, incluso en pacientes con mala adherencia, rara vez se produce el fracaso virológico y, en caso de que esto ocurra, no emergen mutaciones de resistencia7-9,14. Por tanto, uno se siente cómodo y seguro prescribiendo un régimen basado en DRV potenciado, incluso en individuos con riesgo de fracaso virológico. En segundo lugar, es muy buena opción cuando no se dispone del resultado del estudio basal de resistencias o del HLA-B5701 ya que ninguno de los dos son limitantes para el uso de IP.
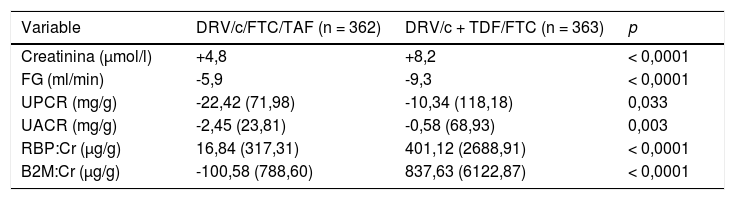
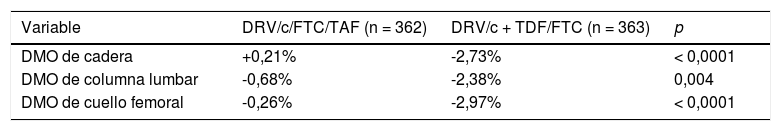
Eficacia y tolerabilidadLa eficacia y seguridad de DRV/c/FTC/TAF (Symtuza®) en pacientes con infección por VIH que no han recibido TAR previamente se ha demostrado en el estudio AMBER15. Ya previamente, un ensayo clínico en fase II, doble ciego, controlado y de distribución aleatoria, se diseñó para comparar la eficacia y seguridad de Symtuza® (n = 103) frente a DRV/c + TDF/FTC (n = 50)16. El principal objetivo fue el porcentaje de pacientes con carga viral (CV) < 50 copias/ml a las 24 semanas por intención de tratar. El 75% de los individuos tratados con Symtuza® y el 74% de DRV/c + TDF/FTC presentaban supresión a las 24 semanas y el 77 y el 84%, respectivamente, a las 48 semanas (diferencia: -6,2%; índice de confianza al 95 % [IC95%]: -19,9-7,4%; p = 0,35); ningún paciente suspendió por fracaso virológico. En el análisis de seguridad, la mayoría de los efectos adversos fueron leves o moderados. Todos los pacientes tenían un filtrado glomerular (FG) basal ≥ 70 ml/min, que disminuyó menos con Symtuza® que con la pauta control (-2,9 frente a -10,6 ml/min; p = 0,017); Symtuza® se asoció, además, con una mejora de los biomarcadores tubulares a las 48 semanas, sin diferencias entre ambas ramas en la proteinuria. En cuanto a la toxicidad ósea, el descenso de la densidad mineral ósea (DMO) tanto en cadera como en columna fue inferior con Symtuza® que con DRV/c + TDF/FTC (p < 0,001 y p = 0,003, respectivamente). En cambio, como cabría esperar, los cambios en el perfil lipídico favorecieron a la rama que incluía TDF.
El AMBER15 es un ensayo clínico en fase III, de distribución aleatoria, controlado y doble ciego, diseñado para evaluar la eficacia y seguridad de Symtuza® frente a DRV/c + TDF/FTC. Se ha llevado a cabo en 121 centros de 10 países (Estados Unidos, Canadá, Bélgica, Francia, Alemania, Italia, Polonia, Rusia, España y Reino Unido). El ensayo incluye una fase de cribado de 4-6 semanas y un período de tratamiento de 48 semanas. Posteriormente, todos los pacientes continúan con Symtuza® en una fase de rama única con apertura del ciego hasta la semana 96 y una fase de extensión. Los individuos se distribuyen aleatoriamente 1:1 para recibir Symtuza® o DRV/c + TDF/FTC, junto con placebo, de tal forma que todos tengan un régimen con 3 comprimidos, y se les instruye para que tomen la medicación con la comida, aproximadamente, a la misma hora por las mañanas. La distribución aleatoria se estratifica por CV basal (≤ o ≥ 100.000 copias/ml) y recuento de linfocitos CD4 (< o ≥ 200 cél./mm3). Todos los participantes son adultos mayores de 18 años, no han recibido TAR previamente, con una CV en el momento de la detección de ≥ 1.000 copias/ml, un recuento de linfocitos CD4 ≥ 50 cél./mm3, sin mutaciones de resistencia para DRV, TDF o FTC, y con un FG basal ≥ 70 ml/min. Los principales criterios de exclusión son el diagnóstico de un episodio relacionado con el sida durante los 30 días previos a la detección, estar coinfectado por el virus de la hepatitis B o C, padecer alguna enfermedad clínicamente importante y estar embarazada o en período de lactancia. Se recogen características epidemiológicas, clínicas y analíticas, incluyendo, además de la CV y recuento de linfocitos CD4, parámetros lipídicos y perfil hepático, renal y óseo.
El fracaso virológico por protocolo (FVDP) se define como la ausencia de respuesta (descenso de < 1 log10 y CV ≥ 50 copias/ml a las 8 semanas, confirmado en la siguiente visita) o un rebote virológico (CV ≥ 50 copias/ml tras haber alcanzado de forma confirmada y consecutiva una CV < 50 copias/ml o bien un incremento de CV ≥ 1 log 10respecto al nadir) o viremia detectable al final de seguimiento (CV ≥ 400 copias/ml al final del estudio o retirada de este tras la semana 8). Se realiza estudio de resistencias después de la detección en las muestras de los pacientes que presentan FVDP y CV ≥ 400 copias/ ml en el momento del fracaso o posteriormente.
El objetivo principal del estudio es evaluar la no inferioridad de Symtuza® frente a DRV/c + TDF/FTC en la proporción de pacientes con CV < 50 copias/ml a 48 semanas y como objetivos secundarios se incluyen la proporción de pacientes con CV < 20 copias/ml, < 200 co-pias/ml, los cambios en la CV y recuento de linfocitos CD4 respecto al valor basal, el desarrollo de resistencias en pacientes con FVDP y la seguridad y tolerancia a las 48 semanas. Para demostrar la no inferioridad, el límite inferior del IC95% de la diferencia en la tasa de respuesta a 48 semanas entre las dos ramas debe ser ≥ 10%.
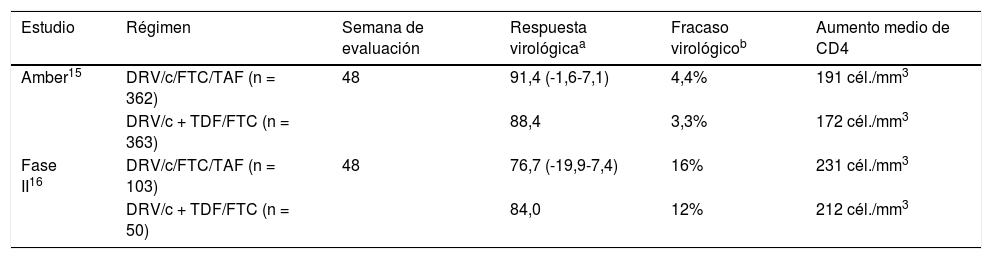
Se distribuye al azar a 725 pacientes, 362 con Symtuza® y 363 en la rama control. Las características basales son similares en ambos grupos, con el 18% de pacientes con CV basal ≥ 100.000 copias/ml y una mediana de recuento de linfocitos CD4 de 453 cél./mm3. El 16% de pacientes presenta basalmente mutaciones de resistencia asociadas con los NNRTI, el 5%, mutaciones de resistencia asociadas con los NRTI y el 2% presenta mutaciones primarias para IP. A las 48 semanas, el 91,4% de pacientes con Symtuza® y el 88,4% de la rama control tiene una CV < 50 copias/ml, con una diferencia de 2,7% (IC95%: -1,6-7,1%; p ≥ 0,0001), lo que demuestra la no inferioridad del STR. Presentan FVDP 8 pacientes en la rama con Symtuza® y 6 pacientes en la rama control, sin emergencia de mutaciones para IP o TDF/FTC. Un paciente desarrolla la 184V. Este paciente tiene la 103N en el genotipo basal y niveles bajos de fármacos en los estudios farmacocinéticos, por lo que previsiblemente el fracaso se asocia con mala adherencia. En la tabla 1 se resume la eficacia virológica de Symtuza® en pacientes que no han recibido TAR previamente en los ensayos en fase II y III.
Eficacia virológica de DRV/c/FTC/TAF (Symtuza®) en pacientes con infección por el VIH que no han recibido tratamiento antirretroviral previamente en ensayos clínicos en fases II y III
| Estudio | Régimen | Semana de evaluación | Respuesta virológicaa | Fracaso virológicob | Aumento medio de CD4 |
|---|---|---|---|---|---|
| Amber15 | DRV/c/FTC/TAF (n = 362) | 48 | 91,4 (-1,6-7,1) | 4,4% | 191 cél./mm3 |
| DRV/c + TDF/FTC (n = 363) | 88,4 | 3,3% | 172 cél./mm3 | ||
| Fase II16 | DRV/c/FTC/TAF (n = 103) | 48 | 76,7 (-19,9-7,4) | 16% | 231 cél./mm3 |
| DRV/c + TDF/FTC (n = 50) | 84,0 | 12% | 212 cél./mm3 |
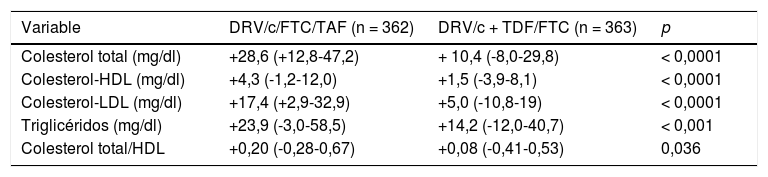
En cuanto al análisis de seguridad, ambos regímenes son muy bien tolerados, con muy escasos efectos adversos de grado 3-4 y sin diferencias en la proporción de individuos que suspendieron el TAR por toxicidad (tabla 2). En cuanto al perfil lipídico, como era previsible, todos los parámetros aumentan más en la rama que incluía TAF que en la de TDF (tabla 2) mientras que el perfil renal y óseo favorecen a Symtuza® (tabla 4 y tabla 5). Ningún paciente de la rama del STR suspende el TAR por efectos adversos renales, óseos o del sistema nervioso central.
Efectos adversos a las 48 semanas
| Incidencia, n (%) | DRV/c/FTC/TAF(n = 362) | DRV/c + TDF/FTC (n = 363) |
|---|---|---|
| ≥ 1 EA de cualquier grado | 312 (86,2) | 307 (84,6) |
| ≥ 1 EA de grado 3-4 | 19 (5,2) | 22 (6,1) |
| ≥ 1 EA grave | 17 (4,7) | 21 (5,8) |
| ≥ 1 EA que causa suspensión | 7 (1,9) | 16 (4,4) |
| Muerte | 0 | 0 |
| EA, al menos, posiblemente relacionados con el fármaco de estudio | ||
| Cualquier EA Más frecuentes (≥ 5% por rama) | 126 (34,8) | 151 (41,6) |
| • Diarrea | 31 (8,6) | 40 (11,0) |
| • Exantema | 22 (6,1) | 14 (3,9) |
| • Náuseas | 20 (5,5) | 36 (9,9) |
Cambio en el perfil lipídico a las 48 semanas respecto al basal
| Variable | DRV/c/FTC/TAF (n = 362) | DRV/c + TDF/FTC (n = 363) | p |
|---|---|---|---|
| Colesterol total (mg/dl) | +28,6 (+12,8-47,2) | + 10,4 (-8,0-29,8) | < 0,0001 |
| Colesterol-HDL (mg/dl) | +4,3 (-1,2-12,0) | +1,5 (-3,9-8,1) | < 0,0001 |
| Colesterol-LDL (mg/dl) | +17,4 (+2,9-32,9) | +5,0 (-10,8-19) | < 0,0001 |
| Triglicéridos (mg/dl) | +23,9 (-3,0-58,5) | +14,2 (-12,0-40,7) | < 0,001 |
| Colesterol total/HDL | +0,20 (-0,28-0,67) | +0,08 (-0,41-0,53) | 0,036 |
Cambio en el perfil renal a las 48 semanas respecto al basal
| Variable | DRV/c/FTC/TAF (n = 362) | DRV/c + TDF/FTC (n = 363) | p |
|---|---|---|---|
| Creatinina (μmol/l) | +4,8 | +8,2 | < 0,0001 |
| FG (ml/min) | -5,9 | -9,3 | < 0,0001 |
| UPCR (mg/g) | -22,42 (71,98) | -10,34 (118,18) | 0,033 |
| UACR (mg/g) | -2,45 (23,81) | -0,58 (68,93) | 0,003 |
| RBP:Cr (μg/g) | 16,84 (317,31) | 401,12 (2688,91) | < 0,0001 |
| B2M:Cr (μg/g) | -100,58 (788,60) | 837,63 (6122,87) | < 0,0001 |
Cambio en el perfil óseo a las 48 semanas respecto al basal
| Variable | DRV/c/FTC/TAF (n = 362) | DRV/c + TDF/FTC (n = 363) | p |
|---|---|---|---|
| DMO de cadera | +0,21% | -2,73% | < 0,0001 |
| DMO de columna lumbar | -0,68% | -2,38% | 0,004 |
| DMO de cuello femoral | -0,26% | -2,97% | < 0,0001 |
En resumen, el AMBER demuestra la no inferioridad de Symtuza® respecto a DRV/c coadministrado con TDF/FTC; el STR se asocia, además, a mejor perfil de seguridad ósea y tubular renal. Symtuza® es el primer STR basado en IP, en el cual a la potencia y alta barrera genética de DRV potenciado se le suma la seguridad y buena tolerancia de TAF, lo que lo sitúa como una opción muy competitiva en pacientes con infección por el VIH que no han recibido TAR previamente.
Por último, el estudio DIAMOND17, cuyos resultados a 24 semanas se acaban de comunicar, demuestra la eficacia y seguridad de Symtu-za® en el inicio inmediato del TAR en pacientes con diagnóstico reciente de infección por el VIH. Este estudio es un ensayo clínico en fase III, de un solo brazo, abierto, prospectivo y multicéntrico para evaluar DRV/c/FTC/TAF en el inicio inmediato del TAR. Todos los participantes son adultos mayores de 18 años, diagnosticados de infección por el VIH durante las 2 semanas previas a la visita basal y no han recibido TAR previamente, salvo el uso de FTC/TDF de profilaxis preexposición. Los principales criterios de exclusión son presentar algún episodio relacionado con el sida que incremente el riesgo de morbimortalidad, padecer alguna hepatopatía o insuficiencia renal crónica con un FG < 50 ml/min. Todos los pacientes inician TAR de forma inmediata, en las primeras 24 horas tras la visita basal, sin disponer de los datos analíticos realizados en esta. A los 3 días (±1 semana) se revisan los parámetros de laboratorio y se suspende el TAR en caso de FG < 50 ml/min, hipertransaminasemia ≥ 2,5 veces el límite superior de la normalidad, lipasa ≥ 1,5 veces el límite superior de la normalidad, test de embarazo positivo o infección activa por VHC que precise tratamiento con fármacos incompatibles con DRV/c/ FTC/TAF. El estudio de resistencias se evalúa a las 4 semanas (±7 días), se retira a los pacientes que no tengan sensibilidad genotípica total a DRV/c/FTC/TAF y solo se permite resistencia a 3TC/FTC asociada con la existencia de la mutación 184I o 184V. El objetivo principal del estudio es la proporción de pacientes con CV < 50 copias/ml a las 48 semanas y como objetivos secundarios se incluyen la proporción de pacientes con CV < 200 copias/ml, los cambios en la CV y recuento de linfocitos CD4 respecto al valor basal, el desarrollo de resistencias en pacientes con FVDP y la seguridad y tolerancia a las 48 semanas. Se evalúa, además, la satisfacción con el TAR a las 4 y 24 semanas mediante un cuestionario específico (HIVTSQs). Se incluye a 109 pacientes, el 23% de ellos con una CV basal ≥ 100.000 copias/ml y el 21% con un recuento de linfocitos CD4 < 200 cél./mm3. La mediana de tiempo desde el diagnóstico hasta la visita basal es 5 (0-14) días y el 29% se incluyen en las primeras 48 horas tras el diagnóstico. A las 24 semanas, 99 (91%) de los pacientes continúan con Symtuza® y 10 (9%) han suspendido el TAR (3 por criterios de seguridad, 3 por pérdida de seguimiento, 2 por retirada del consentimiento, 1 por violación del protocolo y 1 por efectos adversos). Ningún paciente se retira por los resultados del estudio de resistencias basal. La proporción de pacientes con CV < 50 copias/ml a las 24 semanas es del 81% (FDA snapshot) y el 90% (datos observados). No hay ningún FVDP. En cuanto al análisis de seguridad, el régimen se tolera muy bien, con escasos efectos adversos de grado 3-4, y el cuestionario HIVTSQs muestra altos niveles de satisfacción de los pacientes.
¿Qué beneficios muestra Symtuza® en el paciente que no ha recibido tratamiento antirretroviral previamente?En la terapia de inicio, Symtuza® aporta, por un lado, las ventajas o beneficios que se derivan de las características intrínsecas de DRV potenciado y, por otro lado, las derivadas de ir coformulado con TAF en un STR7-9,14,18-20. En relación con las ventajas de DRV potenciado, hay que destacar sus incuestionables eficacia, potencia y alta barrera genética, que lo posicionan como fármaco de elección en individuos con riesgo de desarrollar mutaciones de resistencia, fundamentalmente por mala adherencia10,11. Es, además, un IP con buen perfil de tolerancia tanto a nivel clínico como metabólico, con el cual se tiene una gran experiencia en la práctica clínica diaria7-9. En terapia de inicio tiene, además, la ventaja de que no hay que esperar a disponer del resultado del estudio de resistencias basal, lo que permite un tratamiento precoz. En su coformulación con FTC/TAF aporta, además, los beneficios de seguridad propios de TAF,21,22, la ventaja de no tener que disponer de HLA-B5701 y, por supuesto, las ventajas propias de los STR en cuanto a comodidad y adherencia12,13. En cuanto a desventajas, quizá la más importante es el riesgo de interacciones por su co-formulación con el potenciador cobicistat23, lo que hay que tener en cuenta siempre a la hora de prescribir un régimen de TAR.
En definitiva, Symtuza® es una muy buena opción como TAR de inicio en pacientes en que sea necesario un tratamiento muy precoz sin disponibilidad de genotipo o HLA-B5701, con riesgo de mala adherencia y con escaso potencial de interacciones farmacológicas.
Conflicto de interesesLa autora Cristina Gómez ha recibido honorarios por conferencias de ViiV Healthcare, Gilead Sciences, Merck Sharp and Dohme y Janssen-Cilag.
El autor Jesús Santos ha recibido honorarios por asesoría científica y por conferencias de ViiV Healthcare, Gilead Sciences, Merck Sharp and Dohme y Janssen-Cilag.
La autora Rosario Palacios ha recibido honorarios por asesoría científica y conferencias de ViiV Healthcare, Gilead Sciences, Merck Sharp and Dohme y Janssen-Cilag.
Información sobre el suplementoEste artículo forma parte del suplemento titulado «Darunavir, cobi-cistat, emtricitabina y tenofovir alafenamida coformulados en el tratamiento de la infección por el VIH», que ha sido patrocinado por Janssen.

