




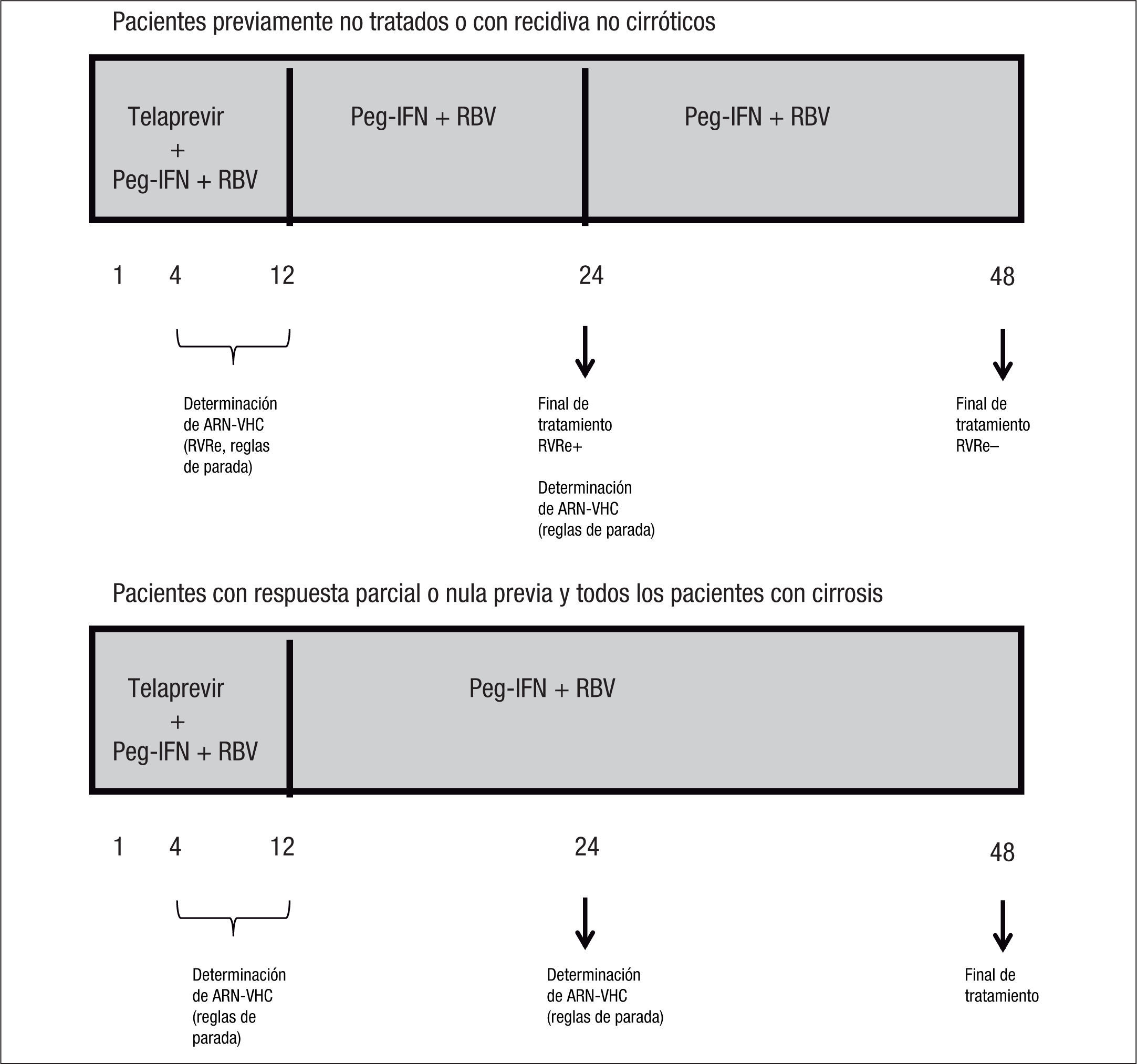
Actualmente, el tratamiento con interferón pegilado, ribavirina y telaprevir se considera el estándar de oro para el tratamiento de la infección crónica por el virus de la hepatitis C. Los aspectos más importantes son el incremento de la tasa de respuesta viral sostenida (el 74–79 frente al 46% de la conseguida con el tratamiento con interferón pegilado y ribavirina) así como la posibilidad de interrupción temprana de una terapia ineficaz gracias a la aplicación de reglas de futilidad a las semanas 4 y 12 (ARN-VHC > 1.000UI/ml), y la posibilidad de seleccionar a los pacientes candidatos a tratamientos más cortos gracias al significado clínico de la respuesta viral rápida extendida (ARN-VHC no detectable a las semanas 4 y 12 de la terapia triple). La duración de la terapia se basa, fundamentalmente, en el estadio de fibrosis y el tipo de respuesta previa con interferón pegilado y ribavirina. Así, tanto para pacientes sin experiencia previa de tratamiento como en aquellos con recidiva con un tratamiento con interferón pegilado, no cirróticos, el tratamiento será de 24 o 48 semanas, dependiendo de la presencia de respuesta viral rápida extendida, mientras que en los pacientes cirróticos y en los respondedores nulos el tratamiento será de 48 semanas.
Los principales efectos adversos durante el tratamiento con telaprevir son la anemia y el exantema cutáneo. Las principales medidas que se deben adoptar ante su presentación son la reducción de la dosis de ribavirina, en el caso de la anemia, y el seguimiento estrecho de las características del exantema con interrupción del tratamiento ante su extensión y su gravedad.
Triple combination therapy with pegylated interferon, ribavirin and telaprevir is currently considered the gold standard for the treatment of chronic hepatitis C virus (HCV) infection. The most important features are an increase in rates of sustained viral response (74–79% vs 46% with pegylated interferon and ribavirin), as well as the possibility of early cessation of ineffective therapy due to the application of futility rules at weeks 4 and 12 (HCV-RNA > 1,000UI/ml), and the possibility of selecting candidates for the shortest treatments due to the clinical significance of extended rapid viral response (undetectable HCV-RNA at weeks 4 and 12 of triple therapy). Treatment length is mainly based on the stage of fibrosis and prior response to pegylated interferon and ribavirin. Thus, in both treatment-naïve patients and patients with recurrence after pegylated interferon therapy, the duration of treatment is 24 or 48 weeks (unless cirrhosis is present), depending on the presence of extended rapid viral response, while in cirrhotic patients and null responders, treatment length is 48 weeks.
The main adverse effects of telaprevir therapy are anemia and skin rash. If these effects occur, the main measures that should be adopted are reduction of the ribavirin dose in anemia, and close monitoring and treatment cessation in skin rash, depending on its spread and severity.
La infección crónica por el virus de la hepatitis C (VHC) constituye la primera causa de cirrosis, carcinoma hepatocelular, trasplante y muerte por causa hepática en el mundo occidental. Existen amplias evidencias de que el aclaramiento del VHC inducido por la terapia antiviral proporciona beneficio con incremento de la supervivencia y disminución de las complicaciones derivadas de la cirrosis1. En los últimos años, los esfuerzos para desarrollar moléculas con acción antiviral directa han resultado en la aprobación reciente de telaprevir y boceprevir, 2 moléculas que actúan inhibiendo la proteasa NS3/4A del VHC genotipo 1, que consiguen aumentar las tasas de respuesta viral sostenida (RVS), tanto en pacientes no tratados previamente como en los que había fallado una pauta previa de tratamiento con interferón pegilado (Peg-IFN) y ribavirina (RBV). Así, en pacientes no tratados previamente infectados por el genotipo 1 del VHC, las tasas de RVS aumentan del 46% con tratamiento estándar al 74–79% si reciben terapia triple con telaprevir, con la ventaja adicional de poder reducir la duración del tratamiento de 48 a 24 semanas en el 63% de los casos2,3. En el caso de pacientes recidivantes al tratamiento previo, las tasas de RVS con terapia triple con telaprevir alcanzan el 84%, con la posibilidad de reducir la duración del tratamiento a 24 semanas en el 70% de los casos. Por último, en los pacientes con respuesta parcial o nula al tratamiento previo, la RVS con terapia triple con telaprevir se consigue en el 61 y 31% de los casos, respectivamente4. Sin embargo, el manejo del tratamiento con estas nuevas moléculas es un importante reto que exige conocer con detalle diversos aspectos que difieren claramente de la terapia estándar con Peg-IFN y RBV, como las reglas de parada, la predicción de la respuesta, la duración de la terapia y el manejo de los efectos adversos. A continuación revisaremos estos aspectos prácticos del uso de la terapia triple con telaprevir.
Reglas de parada o de futilidadEn la terapia triple, tanto con telaprevir como con boceprevir, hay unas reglas de parada o de futilidad que hay que seguir de manera muy estricta en los pacientes que no están respondiendo al tratamiento, a fin de evitar un coste económico innecesario, seguir sometiendo al paciente a posibles efectos adversos sin que aumenten las posibilidades de conseguir RVS y, sobre todo, reducir el riesgo de desarrollo de cepas virales resistentes. En efecto, se sabe que una terapia antiviral inefectiva basada en agentes antivirales directos favorece rápidamente el desarrollo de cepas resistentes. Dado que el VHC existe como cuasiespecies en cualquier persona infectada, con una producción aproximada de 1012 virus al día y aproximadamente 1 error por cada nuevo virus generado, es muy posible que haya cepas virales resistentes antes de iniciarse el tratamiento en casi todos los pacientes, aunque a un nivel bajo5,6. Si estas cepas no son eliminadas rápidamente al iniciar la terapia triple por el Peg-IFN y RBV, continuarán replicando y se convertirán en las cepas dominantes al haber eliminado el inhibidor de la proteasa las cepas sensibles, siendo la consecuencia final inevitable la recidiva viral.
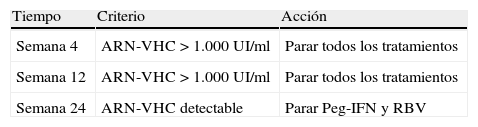
En el caso de la terapia triple con telaprevir, las reglas de parada son presentar un ARN-VHC > 1.000UI/ml a las semanas 4 y 12 de tratamiento, en cuyo caso hay que suspender toda la medicación antiviral, y un ARN-VHC detectable a la semana 24; por tanto, hay que suspender el Peg-IFN y la RBV. De hecho, en un análisis retrospectivo reciente de pacientes tratados con terapia triple con telaprevir en los ensayos clínicos en fase III, se observó que ningún paciente con ARNVHC > 1.000UI/ml a la semana 4 o 12 consiguió RVS a pesar de seguir con el tratamiento7. Es importante, además, que el análisis del ARNVHC sea altamente sensible, debiendo tener un límite inferior de cuantificación ≤ 25UI/ml y un límite de detección de aproximadamente 10–15UI/ml8. En la tabla 1 se muestran las reglas de parada en la terapia triple con telaprevir.
Reglas de parada en la terapia triple con telaprevir.
| Tiempo | Criterio | Acción |
| Semana 4 | ARN-VHC > 1.000UI/ml | Parar todos los tratamientos |
| Semana 12 | ARN-VHC > 1.000UI/ml | Parar todos los tratamientos |
| Semana 24 | ARN-VHC detectable | Parar Peg-IFN y RBV |
Peg-IFN: interferón pegilado; RBV: ribavirina.
El análisis del ARN-VHC debe tener un límite inferior de cuantificación ≤ 25UI/ml y un límite de detección de aproximadamente 10–15UI/ml.
Un elemento importante a considerar antes de iniciar una terapia triple con telaprevir es analizar las posibilidades de respuesta a dicho tratamiento. En efecto, una de las primeras preguntas que se plantean los pacientes cuando se discuten nuevas posibilidades terapéuticas de la hepatitis C es saber cuál es su probabilidad de conseguir erradicar el virus.
Los factores asociados a una probabilidad alta de responder a la terapia triple con telaprevir incluyen la raza, la ausencia de fibrosis avanzada, conseguir una respuesta viral rápida (RVR), conseguir una RVR extendida (RVRe: ARN-VHC no detectable a las semanas 4 y 12 de la terapia triple), el tipo de respuesta a un tratamiento previo con Peg-IFN y RBV (recidiva frente a respuesta parcial o nula al tratamiento previo) y presentar un polimorfismo favorable de la IL28B que, en realidad, es un marcador de la sensibilidad del paciente al IFN. Por el contrario, los factores que se asocian a una menor probabilidad de RVS son la raza negra, la fibrosis avanzada o cirrosis, la infección por el genotipo 1a, la ausencia de RVRe, haber presentado una respuesta nula al tratamiento previo y tener un genotipo desfavorable de la IL28B (T/C o T/T). En los pacientes que presentan tanto factores basales favorables como desfavorables, la posibilidad de conseguir una RVS es más difícil de predecir y viene determinada, fundamentalmente, por la respuesta cinética observada durante el tratamiento.
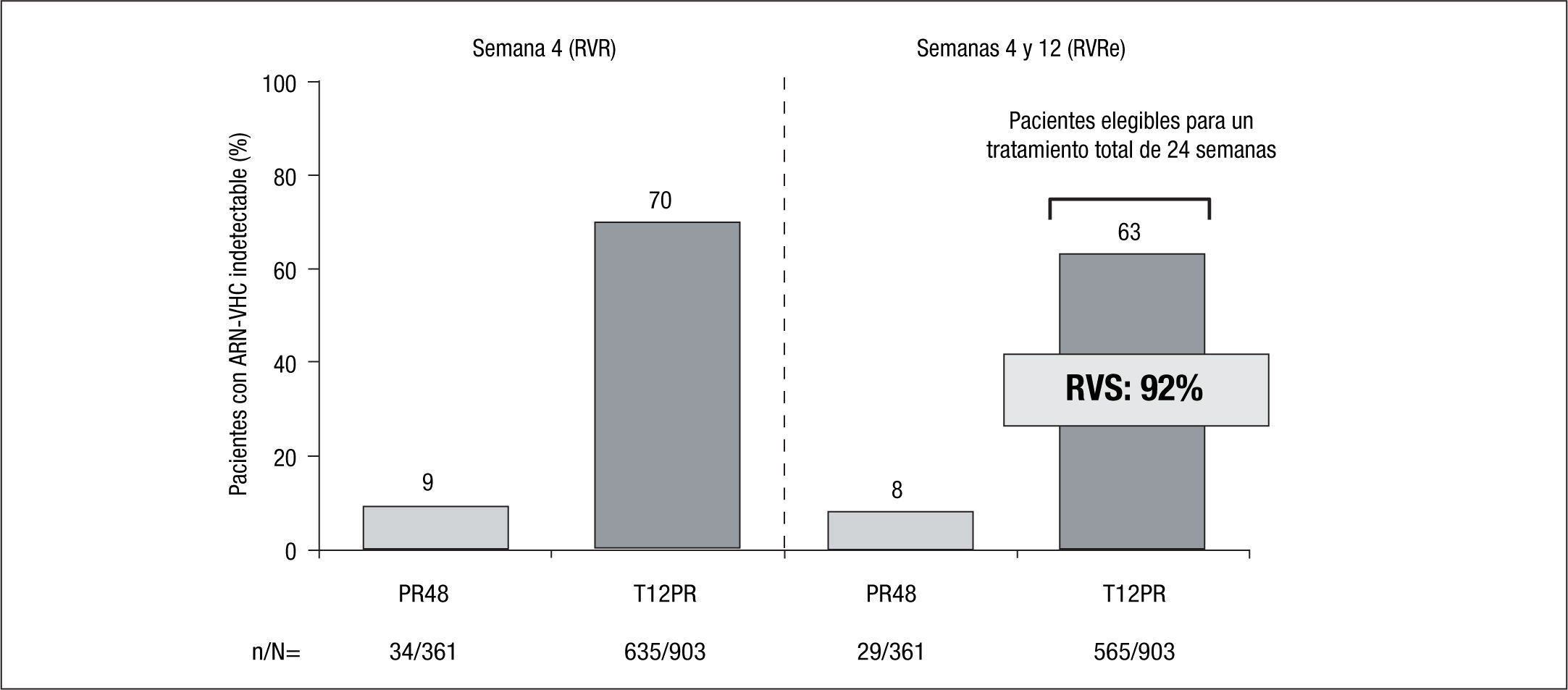
Pacientes no tratados previamenteTanto en el estudio ADVANCE como en el ILLUMINATE2,3, el principal factor predictivo de conseguir una RVS fue la RVRe. En efecto, al agrupar los datos de ambos estudios se observó que el 63% de los pacientes tratados con telaprevir consiguió una RVRe, frente al 8% en los pacientes que recibió terapia estándar. Estos pacientes con RVRe son los susceptibles de acortar el tratamiento a tan solo 24 semanas (salvo que tengan cirrosis) y en ellos se consigue una RVS del 92% (fig. 1). Aunque los pacientes tratados con Peg-IFN/RBV con RVRe consiguen una RVS similar, ello acontece en tan solo una séptima parte de los pacientes, en comparación con la terapia triple. En el estudio ILLUMINATE2 no se observaron diferencias clínicamente significativas en la RVS en los pacientes tratados con telaprevir y que presentaban factores típicos predictores de mala respuesta, como la carga viral basal > 800.000UI/ml, el genotipo 1a o la raza negra en los pacientes que habían conseguido una RVRe.
 no detectable a las semanas 4 y 8 de tratamiento observada en los estudios ADVANCE e ILLUMINATE. PR: interferón pegilado + ribavirina; RVR: respuesta viral rápida; RVRe: respuesta viral rápida extendida; T: telaprevir.")
Otro factor importante que determina la posibilidad de conseguir una RVS es el grado de lesión hepática. En el estudio ADVANCE3, el 78% de los pacientes con ausencia o fibrosis portal consiguió una RVS frente al 47% observado en los pacientes con terapia estándar. En los pacientes con fibrosis en puentes (F3) o cirrosis (F4) tratados con terapia triple con telaprevir, la RVS fue del 62 frente al 33% en los pacientes tratados con terapia estándar. El hecho de que los pacientes con enfermedad avanzada respondan peor al tratamiento apoya la importancia de tratar a los pacientes precozmente para prevenir la progresión de la enfermedad, lo que puede tener un impacto importante en la evolución a largo plazo de estos pacientes.
El polimorfismo de la IL28B del paciente es también un factor predictivo importante de respuesta al tratamiento, consiguiéndose en el estudio ADVANCE unas tasas de RVS con terapia triple con telaprevir del 90, 71 y 73% en los pacientes con genotipos CC, CT y TT, frente al 64, 25 y 23% en los pacientes tratados con terapia estándar, respectivamente. Por otra parte, el polimorfismo de la IL28B informa sobre la posibilidad de acortar el tratamiento basado en telaprevir a las 24 semanas, al conseguir con una mayor frecuencia una RVRe en los pacientes con un genotipo favorable (78, 57 y 45% en los pacientes con genotipo CC, CT y TT, respectivamente).
Pacientes con fracaso al tratamiento previo con interferón pegilado/ ribavirinaEn los pacientes con fracaso al tratamiento previo con Peg-IFN/ RBV, los principales factores predictores de RVS son el tipo de respuesta al tratamiento previo, el grado de fibrosis que presenta el paciente y la negativización rápida de la carga viral durante el tratamiento.
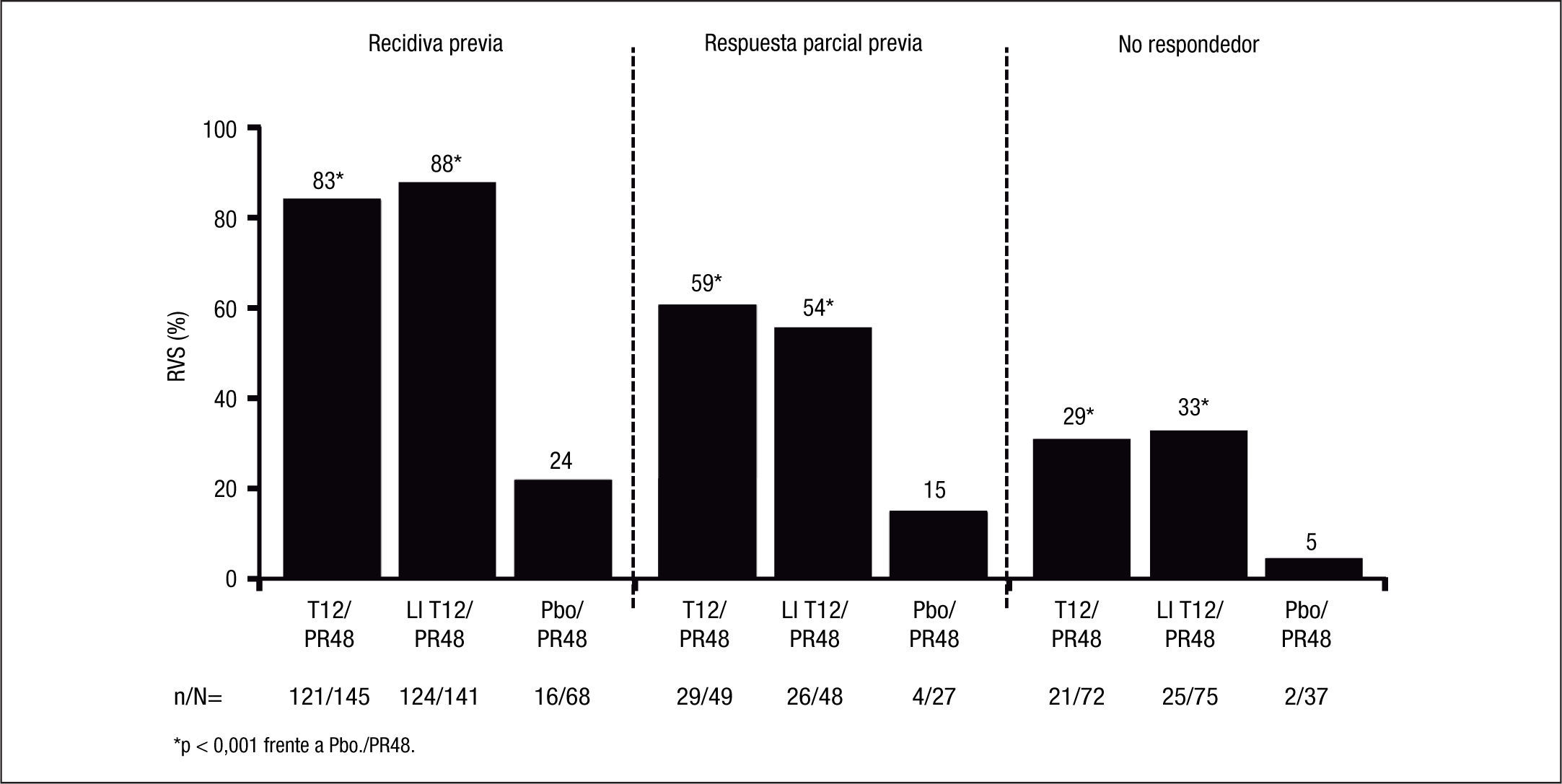
Como era previsible, los pacientes con respuesta nula al tratamiento previo con Peg-IFN/RBV son los más difíciles de tratar, dado que el grado de sensibilidad del paciente al Peg-IFN/RBV, al evitar la emergencia de cepas virales resistentes, condiciona la eficacia de los inhibidores de la proteasa de primera generación, como es telaprevir. En efecto, en un subanálisis del estudio REALIZE4 se evaluaron las tasas de RVS según el tipo de respuesta al tratamiento previo (fig. 2). La posibilidad de conseguir una RVS con terapia triple con telaprevir se relacionó directamente con el tipo de respuesta al tratamiento previo con Peg-IFN/RBV, con RVS del 83 y 88% en los pacientes con recidiva previa, del 54 y 59% en los pacientes con respuesta parcial previa (descenso ≥ 2 log10 en el ARN-VHC respecto al valor basal a la semana 12 de tratamiento, sin lograr que el ARN-VHC fuese no detectable durante el tratamiento) y del 29 y 33% en los pacientes con respuesta nula previa (descenso ≤ 2 log10 en el ARN-VHC respecto al valor basal de la semana 12 de tratamiento). Además, en los pacientes con recidiva previa se consiguió una RVRe en alrededor del 70% de los casos tratados con telaprevir, por lo que en ellos se puede reducir la duración del tratamiento a 24 semanas, a menos que el paciente presente cirrosis.
 observadas en el estudio REALIZE según el tipo de respuesta al tratamiento previo. Pbo: placebo; PR: interferón pegilado + ribavirina; T: telaprevir.")
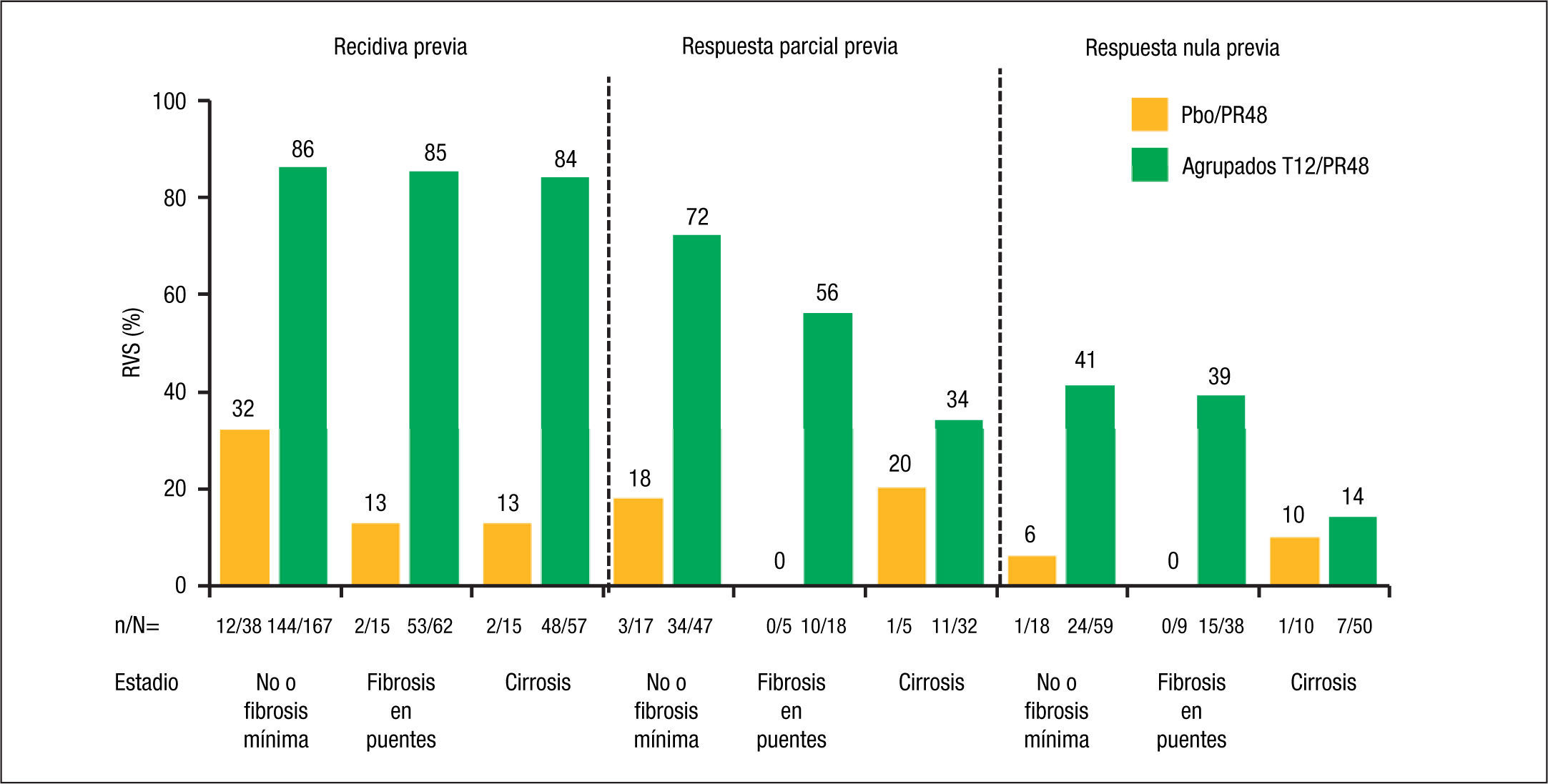
El impacto del grado de fibrosis que presenta el paciente tiene también una gran influencia en la posibilidad de conseguir RVS en los pacientes con un fracaso al tratamiento previo con Peg-IFN/RBV, especialmente en los que presentaron una respuesta parcial o nula previa. Así, en un subanálisis del estudio REALIZE9 se observó que mientras que el grado de fibrosis no influyó en la RVS de los pacientes con recidiva previa (el 84% de RVS en los pacientes con cirrosis tratados con terapia triple con telaprevir), la RVS en los pacientes con cirrosis descendió al 34 y al 14% en los pacientes con respuesta parcial y nula previas, respectivamente (fig. 3). Hay que tener en cuenta, no obstante, que el número de pacientes estudiados en algunos grupos de este subanálisis es muy pequeño, por lo que estos datos deben tomarse con cautela a la espera de disponer de resultados en estudios con un número más amplio de pacientes.
 observadas en el estudio REALIZE según el tipo de respuesta al tratamiento previo y el grado de fibrosis. Pbo: placebo; PR: interferón pegilado + ribavirina; T: telaprevir.")
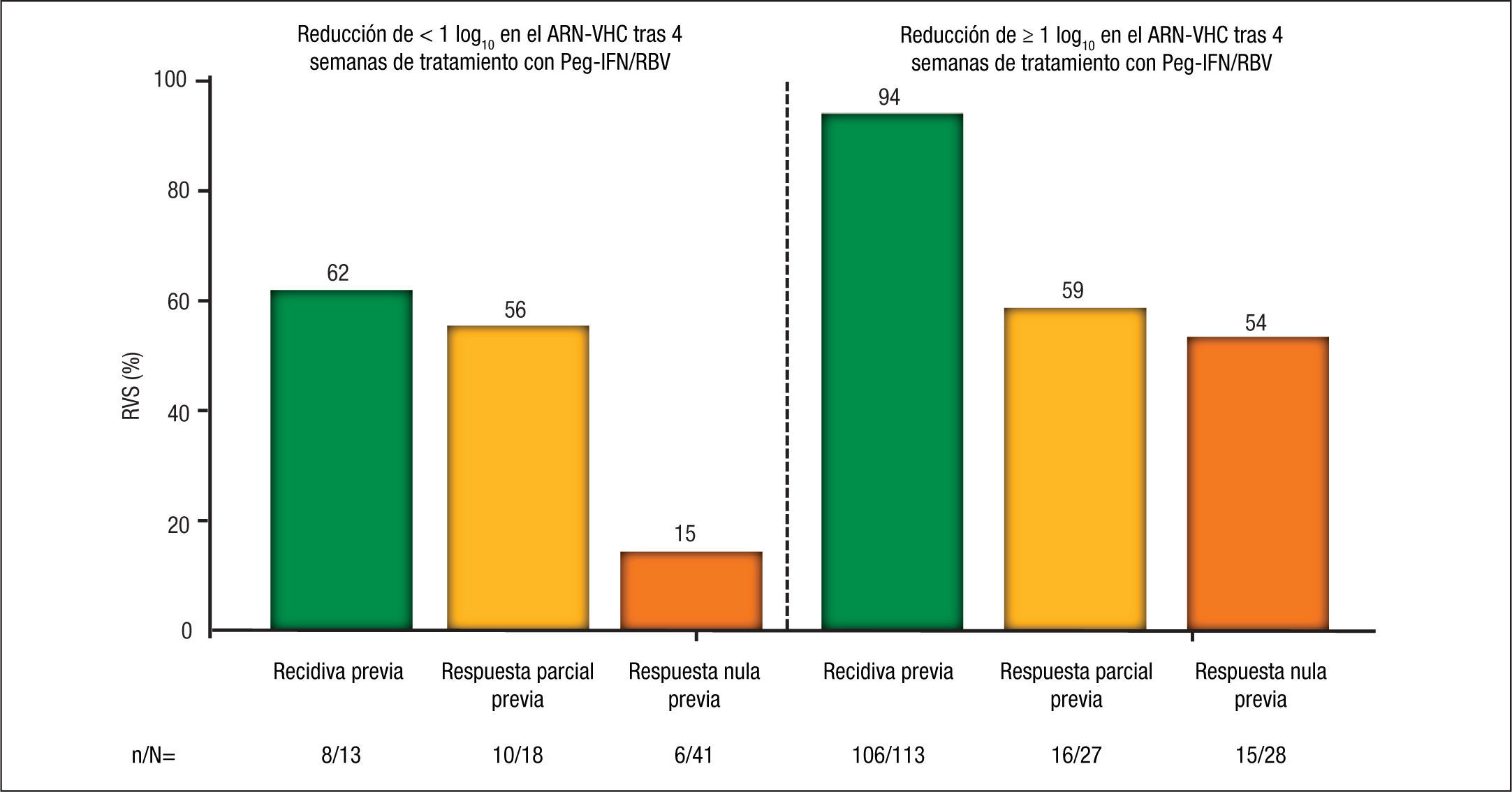
Por último, la negativización rápida de la carga viral durante el tratamiento también constituye un factor predictor de RVS en los pacientes con fracaso al tratamiento previo. En uno de los dos brazos con terapia con telaprevir del estudio REALIZE se evaluó la pauta de “leadin” (tratamiento inicial con Peg-IFN y RBV durante las primeras 4 semanas). Aunque no se observó beneficio en la tasa de respuesta durante el tratamiento, rebote viral o RVS en los pacientes que recibieron una pauta de “lead-in” previa a la introducción del telaprevir, esta fase demostró otras ventajas, como ser un importante factor predictivo de respuesta viral, obteniéndose una tasa de RVS global del 81% en los pacientes que consiguieron un descenso ≥ 1 log10 tras el “lead-in”, frente al 33% en los que no lo consiguieron. Esta fase de “lead-in” es especialmente importante, tal y como aconsejan varios documentos de recomendaciones10,11, tanto en los pacientes en los que se desconoce el tipo de respuesta al tratamiento previo, en los que esta fase nos permitiría conocer en estos pacientes la sensibilidad al IFN antes de la introducción del telaprevir, como en aquellos con respuesta nula al tratamiento previo, dado que la posibilidad de RVS es tan solo del 15% si la reducción del ARN-VHC es < 1 log10 a las 4 semanas de la fase “lead-in” frente al 54% si la reducción es ≥ 1 log10 (fig. 4).
Duración de la terapia![Tasas de respuesta viral sostenida (RVS) observadas en el brazo “lead-in” (4 semanas de tratamiento con interferón pegilado [Peg-IFN]/ribavirina [RBV]) del estudio REALIZE según la reducción del ARN-VHC (virus de la hepatitis C) a la semana 4 de tratamiento.](https://static.elsevier.es/multimedia/0213005X/00000031000000S3/v1_201309220012/S0213005X13701204/v1_201309220012/es/main.assets/gr4.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNc+34VOwP+2HTB5hao7CSfc3vHAcopEDD4TvJ2qcebX0k9HqN8G7zZXrpLvzzF4z81M6oqnjs5m0zKSRtcWqikyRyGrv89Wx4zUscpjT4kKocOdSAyYGtaJHJ8IYyssLsdBq+RVJx6Oblhsa8l7aUQWiLEr7riZkHtZ5G+syps7Hzfx24CoRpcVKGRIltJU3wT4vzfsh5Q4tp1+NstUnqRu9nxIRDJmrmdhyTlwfivKgOx8Z4wQG+30CzTuQ7EevSa1YAGIfaVvBX8iy5H3WOh0 "Tasas de respuesta viral sostenida (RVS) observadas en el brazo “lead-in” (4 semanas de tratamiento con interferón pegilado [Peg-IFN]/ribavirina [RBV]) del estudio REALIZE según la reducción del ARN-VHC (virus de la hepatitis C) a la semana 4 de tratamiento.")
Tal y como se ha comentado en los apartados previos, el fundamento para establecer la duración de la terapia combinada con telaprevir se basa en la existencia de factores predictivos de aclaramiento viral con este tratamiento, que permiten a un determinado grupo de pacientes recibir tratamientos más cortos en beneficio de una menor exposición a efectos secundarios y ahorro económico. A pesar de que son varios los factores predictivos de respuesta al tratamiento con telaprevir, en la actualidad, la duración del tratamiento será distinta según la RVRe, el estadio de fibrosis (cirrosis/no cirrosis) y la respuesta a esquemas previos con IFN y RBV.
La importancia de la RVRe en la toma de decisiones en cuanto a la duración del tratamiento triple que incluye telaprevir (duración guiada por la respuesta: response guided therapy, RGT) tiene su primera evidencia en los resultados del estudio ADVANCE3, en el que los pacientes previamente no tratados de los brazos de tratamiento que incluían telaprevir y consiguieron RVRe (RVRe+) se trataron 24 semanas, mientras que los que no consiguieron RVRe (RVRe–) se trataron 48 semanas. La tasa de RVS en el brazo de tratamiento con telaprevir fue del 73%, siendo del 89% en los que se trataron 24 semanas (RVRe+) y del 54% en los que se trataron 48 semanas (RVRe–). De este estudio es importante destacar que se estableció que hasta un 58% de los pacientes eran candidatos a recibir tratamiento durante 24 semanas (fig. 1). El estudio ILLUMINATE2 estableció definitivamente la duración del tratamiento combinado con telaprevir en los pacientes previamente no tratados, ya que demostró la no inferioridad del tratamiento durante 24 semanas frente al tratamiento durante 48 semanas en los pacientes con RVRe+. Sin embargo, la duración del tratamiento que se establece a partir de los resultados de estos estudios tiene una salvedad, y es en el paciente cirrótico. Por un lado, tanto en el estudio ADVANCE3 como en el estudio ILLUMINATE2 se incluyó una proporción muy baja de cirróticos (el 7 y el 11% en los estudios ADVANCE3 e ILLUMINATE2, respectivamente), lo que impide la emisión de recomendaciones en esta población y, por otro, datos del estudio ILLUMINATE2 indican que la RVS en los pacientes que recibieron tratamiento durante 24 semanas, a pesar de haber alcanzado RVRe, era del 61 frente al 92% del brazo de tratamiento durante 48 semanas. De todo ello se establece que la duración del tratamiento será de 24 (RVRe+) o 48 semanas (RVRe–) en todos los pacientes previamente no tratados, excepto en los cirróticos, en los que la duración del tratamiento deberá ser de 48 semanas con independencia de la RVRe.
La eficacia del tratamiento combinado con telaprevir en función de la respuesta a tratamientos previos con Peg-IFN y RBV se basa en los datos del estudio REALIZE4. En este estudio se demostró que la RVS en los pacientes con recidiva previa (ARN-VHC detectable durante el seguimiento cuando era indetectable al final del tratamiento), respuesta parcial (disminución ≥ 2 log ARN del VHC a las 12 semanas de tratamiento con ARN detectable al final del tratamiento) y respuesta nula (disminución < 2 log ARN del VHC a las 12 semanas de tratamiento) era del 88, 59 y 29%, respectivamente, con una disminución hasta del 15% en los pacientes cirróticos con respuesta previa nula (figs. 2 y 3). Así pues, se ha establecido que la duración del tratamiento deberá ser de 24 semanas en los pacientes con recidiva previa que consigan RVRe+ y de 48 semanas en los pacientes con recidiva y RVRe–, así como en los pacientes con respuesta previa parcial o nula.
En la figura 5 se resume la duración del tratamiento en función de la RVRe, histología hepática y respuesta a tratamientos previos. Asimismo se incluyen las determinaciones de ARN-VHC para la aplicación de las reglas de parada y la definición de RVRe.
Manejo de los efectos adversos, histología hepática y patrón de respuesta a esquemas previos de tratamiento. Peg-IFN: interferón pegilado; RBV: ribavirina.")
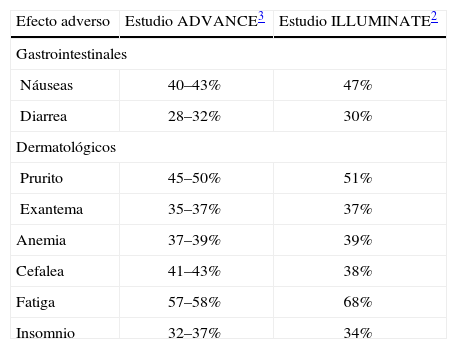
La aparición de efectos adversos de diversa índole durante la terapia con telaprevir alcanza al 99% de los pacientes, aunque únicamente un 9% presenta efectos adversos graves. En la tabla 2 se señalan los principales efectos adversos observados en los estudios ADVANCE3 e ILLUMINATE2. Sin embargo, a pesar de la alta incidencia de efectos adversos, un porcentaje muy bajo de pacientes interrumpió este tratamiento debido a efectos adversos. Concretamente, en el estudio ILLUMINATE únicamente el 12% abandonó el tratamiento de telaprevir por efectos adversos (el 18% abandonó los 3 fármacos por efectos adversos), mientras que el porcentaje total de abandonos ascendió al 34%. Porcentajes similares se han comunicado en los estudios ADVANCE3 y REALIZE4.
Principales efectos adversos de los estudios ADVANCE3 e ILLUMINATE2
De todos los efectos descritos durante el tratamiento con telaprevir, sin duda los más importantes son la anemia, el exantema cutáneo y los síntomas anorrectales.
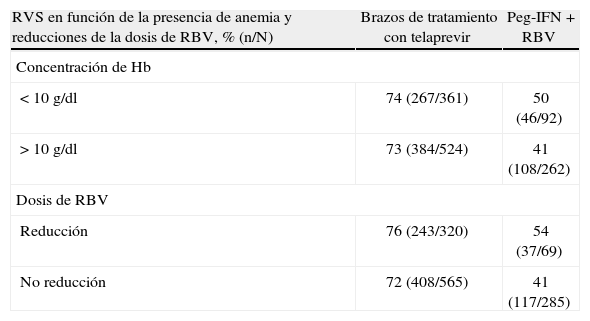
Tal como puede observarse en la tabla 2, casi un 40% de los pacientes que reciben telaprevir presenta disminución de la concentración de hemoglobina, aunque un porcentaje muy inferior presenta una concentración de hemoglobina ≤ 10g/dl (el 27 y el 11% entre 10–8,5 y < 8,5g/dl, respectivamente)4. Es importante destacar que la anemia aparece en general a las 4 semanas del inicio de telaprevir y desaparece progresivamente con retorno de la concentración de hemoglobina a sus valores basales al interrumpir el tratamiento con telaprevir. La anemia fue la causa de interrupción del tratamiento únicamente en el 1% de los pacientes del estudio REALIZE. El diseño de este estudio no permitió la administración de eritropoyetina para el manejo de la anemia, por lo que los pacientes que presentaron reducciones de la concentración de hemoglobina sufrieron reducciones escalonadas de dosis de RBV de acuerdo con la ficha técnica, procediéndose a la interrupción de telaprevir ante la persistencia de la anemia a pesar de la reducción de la dosis o retirada de la RBV. En el estudio ILLUMINATE, cuyo diseño sí permitía la administración de eritropoyetina e incluso la transfusión de concentrados de hematíes, la aparición de anemia grave se presentó en el 6% de los pacientes, 32 pacientes recibieron transfusiones sanguíneas y 7 recibieron eritropoyetina. Subestudios posteriores de los estudios ILLUMINATE3 y ADVANCE2 han demostrado que la reducción de la dosis de RBV no tiene efecto negativo sobre la tasa de RVS (tabla 3)13. En la práctica clínica diaria, no hay contraindicación para la administración de eritropoyetina en los pacientes que presenten anemia durante el tratamiento con telaprevir, pero hay que recordar que el tratamiento de la anemia en esta situación no es una indicación aprobada para el uso de eritropoyetina.
Respuesta viral sostenida en los pacientes de los estudios ADVANCE3 e ILLUMINATE2 en función de la presencia de anemia y de las reducciones de ribavirina (RBV).
| RVS en función de la presencia de anemia y reducciones de la dosis de RBV, % (n/N) | Brazos de tratamiento con telaprevir | Peg-IFN + RBV |
| Concentración de Hb | ||
| < 10g/dl | 74 (267/361) | 50 (46/92) |
| > 10g/dl | 73 (384/524) | 41 (108/262) |
| Dosis de RBV | ||
| Reducción | 76 (243/320) | 54 (37/69) |
| No reducción | 72 (408/565) | 41 (117/285) |
Hb: hemoglobina; Peg-IFN: peginterferón; RVS: respuesta viral sostenida.
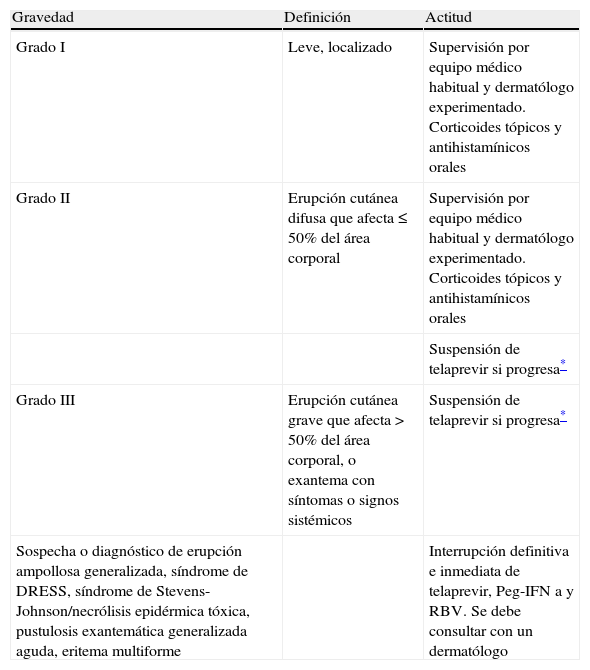
El exantema o rash cutáneo es un efecto adverso importante no solo por su frecuencia de presentación durante el tratamiento con telaprevir, sino por su gravedad en determinados casos. Tal como señala la tabla 2, hasta un 37% de pacientes presenta algún tipo de exantema durante el tratamiento con telaprevir, pero su presencia fue la causa de interrupción del tratamiento combinado con telaprevir únicamente en el 1% de los pacientes del estudio REALIZE4. El exantema típico de telaprevir suele aparecer dentro de las primeras 4 semanas de tratamiento, aunque puede aparecer dentro de las 12 semanas de tratamiento y mejora con la interrupción de este fármaco aunque puede durar semanas. Es, en general, una erupción eccematosa generalizada y solo un porcentaje < 1% presenta reacciones cutáneas adversas graves como el exantema con eosinofilia y síntomas sistémicos (síndrome de DRESS) o síndrome de Stevens-Johnson. El manejo del exantema asociado al tratamiento con telaprevir requiere la participación del paciente, personal sanitario a cargo del paciente mientras está siendo tratado y el asesoramiento de un dermatólogo experto. Se debe entrenar al paciente a comunicar al equipo sanitario la presencia de prurito, cualquier tipo de exantema, úlceras mucosas, inyección conjuntival, fiebre o edema facial. Para valorar la gravedad del exantema debe valorarse tanto la extensión inicial como su progresión los días siguientes a su inicio. En la tabla 4 se especifican los pasos a seguir ante la presencia de exantema y su gravedad. En cuanto se ha procedido a la interrupción de telaprevir, este no puede reintroducirse ni tampoco deben reducirse las dosis de telaprevir.
Manejo del exantema cutáneo secundario al tratamiento con telaprevir en función de la gravedad (extraído de la ficha técnica de Incivo8).
| Gravedad | Definición | Actitud |
| Grado I | Leve, localizado | Supervisión por equipo médico habitual y dermatólogo experimentado. Corticoides tópicos y antihistamínicos orales |
| Grado II | Erupción cutánea difusa que afecta ≤ 50% del área corporal | Supervisión por equipo médico habitual y dermatólogo experimentado. Corticoides tópicos y antihistamínicos orales |
| Suspensión de telaprevir si progresa* | ||
| Grado III | Erupción cutánea grave que afecta > 50% del área corporal, o exantema con síntomas o signos sistémicos | Suspensión de telaprevir si progresa* |
| Sospecha o diagnóstico de erupción ampollosa generalizada, síndrome de DRESS, síndrome de Stevens-Johnson/necrólisis epidérmica tóxica, pustulosis exantemática generalizada aguda, eritema multiforme | Interrupción definitiva e inmediata de telaprevir, Peg-IFN a y RBV. Se debe consultar con un dermatólogo |
DRESS: drug reaction, eosinophilia and systemic symptoms; Peg-IFN: interferón pegilado; RBV: ribavirina.
Los síntomas anorrectales se presentan hasta en el 28% de los pacientes que reciben telaprevir4, aunque son una causa infrecuente de abandono del tratamiento (únicamente 2 pacientes abandonaron telaprevir y 3 abandonaron el estudio REALIZE4 debido a síntomas anorrectales). Los síntomas más frecuentes son prurito, hemorroides y proctalgia.
Conflicto de interesesCristina Tural declara formar parte del consejo asesor nacional de Janssen.
Ramón Planas declara haber participado como ponente y haber recibido ayudas de Roche Farma, Janssen, Gilead, BMS y MSD.

