




La extraordinaria potencia antiviral de telaprevir puede verse comprometida por la rápida selección de mutaciones de resistencia, igual que ocurre con otros antivirales de acción directa (AAD) frente al virus de la hepatitis C (VHC). La elevada tasa de replicación del VHC, la ausencia de actividad correctora de errores de su polimerasa, una barrera genética baja para la resistencia o una mala adherencia a la terapia puede favorecer este fenómeno. Telaprevir, al igual que el resto de inhibidores de la proteasa frente al VHC, presenta una barrera genética baja para la resistencia. Los ensayos clínicos en fases II/III (ADVANCE, ILLUMINATE y REALIZE) han caracterizado genotípica y fenotípicamente la resistencia a telaprevir en combinación con interferón pegilado/ribavirina. Las mutaciones se seleccionan principalmente en las posiciones 36, 54, 55, 155 y 156, siendo el perfil de resistencias dependiente del subtipo genético del VHC, selección de mutaciones en las posiciones 36 y 155 para el subtipo 1a y en las posiciones 36, 54, 55, y 156 para el subtipo 1b. La combinación V36M+R155K y los cambios A156F/T/V confieren alto grado de resistencia (FC>25), mientras que el impacto en la resistencia de otras mutaciones como V36A/G/M, T54A/S, R155G/K/M/T y A156S es menor (FC 3-25). La presencia de polimorfismos basales o mutaciones asociadas con resistencia a telaprevir es muy baja (< 1%), especialmente para mutaciones con mayor impacto en la resistencia. Hay un alto grado de resistencia cruzada entre los inhibidores de la proteasa. Sin embargo, la rápida reversión de mutaciones de resistencia a telaprevir, una vez finalizado el tratamiento, podría permitir el uso de estrategias de “reciclaje” con inhibidores de la proteasa.
Telaprevir is an extremely potent antiviral drug. However, as with other direct-acting antiviral agents against the hepatitis C virus (HCV), this potency can be compromised by the rapid emergence of resistance mutations. This phenomenon is favored by the high rate of HCV replication, the lack of corrective activity to HCV polymerase errors, a low genetic barrier to resistance, and poor treatment adherence. Like other HCV protease inhibitors, telaprevir has a low genetic barrier to resistance. Phase II/III clinical trials (ADVANCE, ILLUMINATE and REALIZE) have characterized resistance to telaprevir in combination with pegylated interferon and ribavirin both genotypically and phenotypically. Mutations are selected mainly in positions 36, 54, 55, 155 and 156. The resistance profile depends on the genetic subtype of HCV, with selection of mutations in positions 36 and 155 for subtype 1a and in positions 36, 54, 55 and 156 for subtype 1b. The V36M+R155K and A156F/T/V variants confer a high degree of resistance (>25-fold increase), while the impact on resistance of other mutations such as V36A/G/M, T54A/S, R155G/K/M/T and A156S is lower (3-25- fold increase). The presence of polymorphisms or mutations associated with telaprevir resistance is very low (< 1%), especially for mutations with a higher impact on resistance. There is a high degree of cross resistance among protease inhibitors. However, the rapid reversion of telaprevir resistance mutations after the end of treatment could allow the use of “recycling” strategies with protease inhibitors.
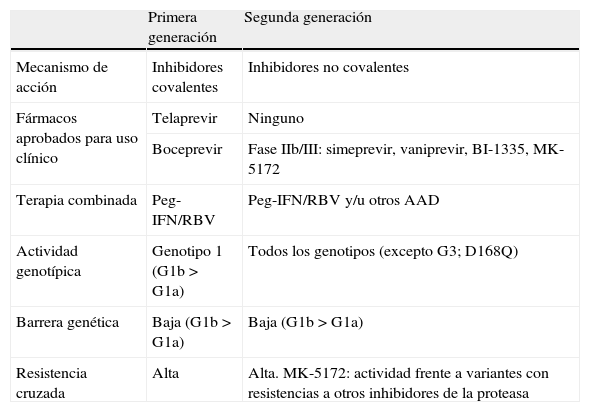
Los inhibidores de la proteasa, telaprevir y boceprevir, han sido los primeros agentes antivirales de acción directa (AAD) aprobados para el tratamiento de la infección crónica por el virus de la hepatitis C (VHC) con genotipo 1 en combinación con el tratamiento estándar interferón pegilado y ribavirina (Peg-IFN/RBV). Se trata de derivados lineales α-cetoamida que se unen covalentemente y de forma reversible a la proteasa NS3/4A del VHC con una cinética de unión y disociación lenta. Ambos incrementan de forma significativa las tasas de curación y, en general, reducen el tiempo de duración del tratamiento1–5. A pesar de la extraordinaria potencia antiviral demostrada por estos fármacos, las elevadas tasas de replicación en la infección por VHC y la baja fidelidad de copia de su polimerasa pueden favorecer la rápida emergencia de mutaciones de resistencias. Dichas mutaciones se seleccionan principalmente cerca del dominio catalítico de la proteasa del VHC. Los inhibidores de la proteasa frente al VHC presentan una barrera genética baja para la resistencia, de manera que la selección de una única mutación puede comprometer la actividad de estos fármacos. Telaprevir y boceprevir tienen un perfil de resistencia muy similar y estudios in vitro han demostrado que hay un alto grado de resistencia cruzada entre ellos6–8 (tabla 1).
Principales características de la resistencia a los inhibidores de la proteasa.
| Primera generación | Segunda generación | |
| Mecanismo de acción | Inhibidores covalentes | Inhibidores no covalentes |
| Fármacos aprobados para uso clínico | Telaprevir | Ninguno |
| Boceprevir | Fase IIb/III: simeprevir, vaniprevir, BI-1335, MK-5172 | |
| Terapia combinada | Peg-IFN/RBV | Peg-IFN/RBV y/u otros AAD |
| Actividad genotípica | Genotipo 1 (G1b > G1a) | Todos los genotipos (excepto G3; D168Q) |
| Barrera genética | Baja (G1b > G1a) | Baja (G1b > G1a) |
| Resistencia cruzada | Alta | Alta. MK-5172: actividad frente a variantes con resistencias a otros inhibidores de la proteasa |
AAD: antivirales de acción directa; Peg-IFN: interferón pegilado.
La mayor parte de los datos disponibles de resistencias a inhibidores de la proteasa del VHC proceden de estudios de pacientes tratados con telaprevir. Los resultados de los ensayos clínicos en monoterapia con telaprevir en pacientes naïve demostraron la rápida aparición de mutaciones (7–10 días) que comprometen la actividad antiviral del fármaco. Dichos estudios permitieron caracterizar in vivo el perfil de resistencias genotípicas y fenotípicas a telaprevir, así como evaluar el impacto de las mutaciones de resistencias en la capacidad replicativa del VHC. Además se pudo comprobar que una vez finalizado el tiempo de tratamiento, la mayor parte de la población viral portadora de mutaciones de resistencias era reemplazada por una población viral salvaje en un tiempo aproximado de entre 3 y 7 meses9. Posteriormente se pudo caracterizar la resistencia a telaprevir en terapia triple en combinación con Peg-IFN/RBV en las fases II/ III de los ensayos clínicos con telaprevir ADVANCE e ILLUMINATE (pacientes naïve) y REALIZE (pacientes pretratados)3–5,10,11.
En este capítulo se revisan las principales características de la resistencia a telaprevir considerando aspectos virológicos, farmacológicos y clínicos.
Selección de resistencias a los agentes antivirales de acción directa en la infección por virus de la hepatitis CLa selección de resistencias a los AAD es el resultado de una incompleta supresión de la replicación viral en presencia de estos fármacos. Hay diversos factores que pueden influenciar este fenómeno, como la alta tasa de replicación del VHC, la baja fidelidad de copia y ausencia de actividad correctora de errores de su polimerasa, una potencia antiviral subóptima del fármaco, una barrera genética baja para la resistencia y una mala adherencia del paciente a la terapia.
Se estima que en un individuo infectado por VHC, la producción viral es del orden de 1012 viriones por día. Esta elevada tasa de replicación sumada a la baja fidelidad de copia y ausencia de actividad correctora de errores de la ARN polimerasa, ARN dependiente del VHC, favorece la generación de una población viral heterogénea formada por genomas virales parecidos pero no idénticos, conocidos como cuasiespecies (tasa de error estimada de 10-3 a 10-5 mutaciones por nucleótido por genoma replicado)12. Dentro de esta población heterogénea puede haber diferentes variantes del VHC que presentan mutaciones específicas asociadas con resistencias a los AAD y que pueden ser seleccionadas positivamente bajo presión farmacológica. Así, en un individuo pueden preexistir variantes del VHC portadoras de cambios asociados con resistencia a los diferentes AAD antes del inicio del tratamiento. En ausencia de una supresión viral completa, dichas variantes pueden seleccionarse y hacerse predominantes en la población viral durante la exposición a los AAD, provocando el fracaso terapéutico. Por el contrario, cuando se consigue una inhibición completa de la replicación del VHC, el escape y enriquecimiento de la población viral con variantes resistentes no es posible13–15.
Con excepción de los inhibidores de la polimerasa análogos de nucleósido, los AAD se caracterizan por presentar una barrera genética baja para el desarrollo de resistencias, es decir, la actividad antiviral de estos fármacos puede verse comprometida por la selección de una única mutación en el genoma viral. De hecho, en fases iniciales de los ensayos clínicos se ha observado cómo el uso de algunos AAD en monoterapia puede favorecer la rápida selección de resistencias en el VHC y el fracaso terapéutico. Por el contrario, la combinación de diferentes agentes antivirales que bloquean diferentes etapas del ciclo de replicación podría ser la mejor estrategia para evitar la selección de mutaciones de resistencias y asegurar el éxito terapéutico, como ocurre en el tratamiento de la infección por el virus de la inmunodeficiencia humana (VIH)6,7.
Como en el caso de otros virus ARN, la infección por VHC se caracteriza por presentar una extraordinaria variabilidad genética, incluso más pronunciada que en el caso de la infección por VIH. Hasta el momento se han descrito 6 genotipos y dentro de ellos diferentes subtipos con una variabilidad global a nivel nucleotídico de hasta un 25%. Se pueden observar diferencias importantes entre la polimerasa, la proteasa y la proteína NS5A entre las diferentes variantes del VHC, de manera que determinados polimorfismos genéticos pueden ser frecuentes en algunas variantes virales y estar ausentes en otras16,17. Algunos de estos polimorfismos pueden encontrarse en posiciones asociadas con la susceptibilidad a los diferentes AAD. En este sentido se han realizado diferentes estudios para evaluar la prevalencia de polimorfismos basales asociados con resistencias a los AAD en pacientes infectados por diferentes genotipos/subtipos del VHC en pacientes naïve. Estos estudios han demostrado que la prevalencia de polimorfismos naturales asociados con resistencias a los diferentes AAD varía en función del genotipo y del subtipo genético del VHC, de manera que la actividad antiviral de los AAD podría ser dependiente del genotipo/subtipo18–25. La relevancia de este fenómeno se ha demostrado en ensayos clínicos que han evaluado la eficacia y seguridad de inhibidores de la proteasa, inhibidores de la polimerasa noanálogos de nucleósido e inhibidores de la proteína NS5A. En ellos, los pacientes infectados con el genotipo 1a del VHC son los que experimentan un mayor número de fracasos terapéuticos y presentan una barrera genética más baja para la resistencia que los pacientes infectados con el genotipo 1b3–5,26–29. Además, los patrones de mutaciones de resistencias pueden variar en función de los genotipos y subtipos genéticos del VHC debido a la variabilidad genética existente.
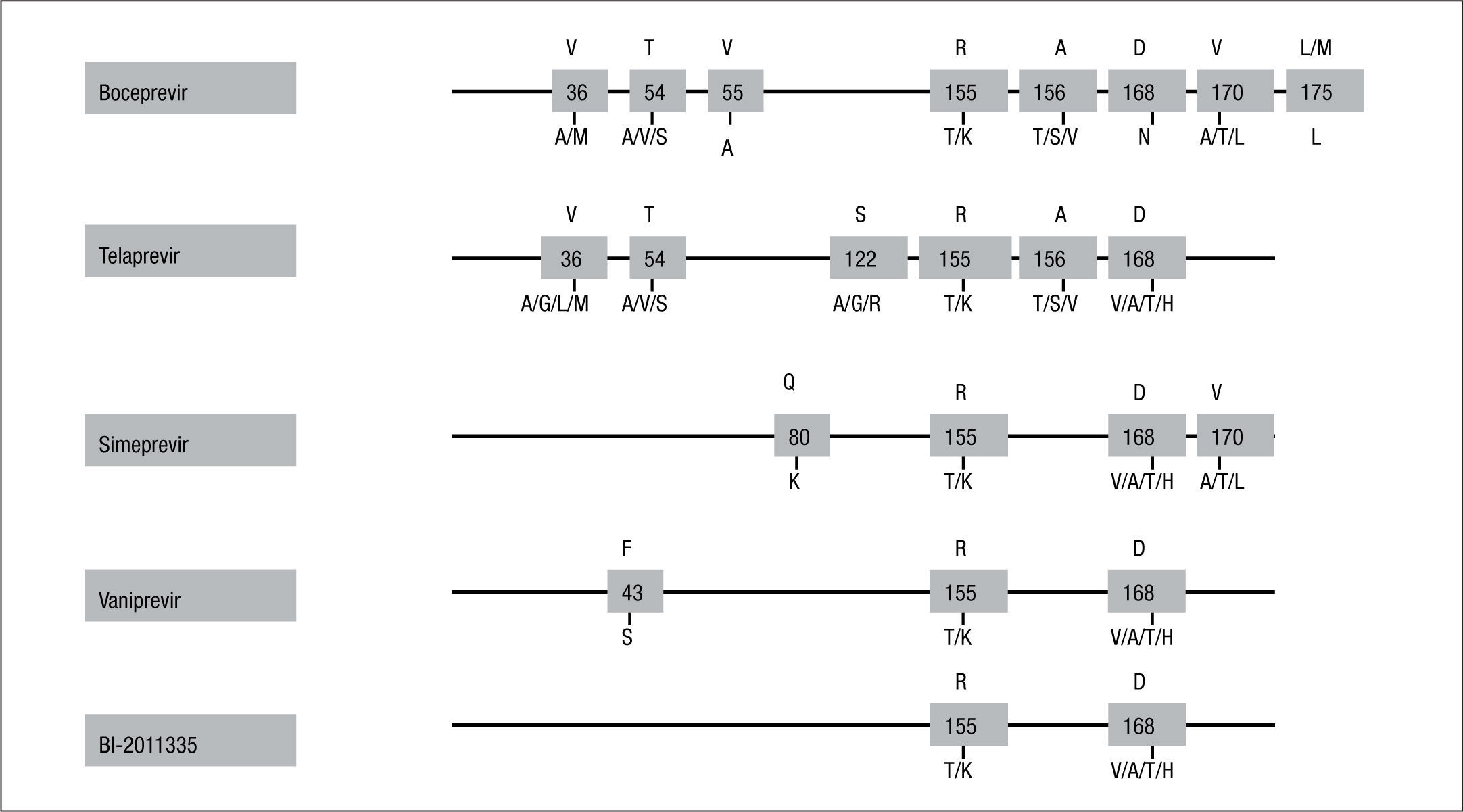
Patrones de resistencias a los inhibidores de la proteasa del virus de la hepatitis CHasta el momento se han identificado diferentes cambios en distintas posiciones de la proteasa del VHC asociados con pérdida de susceptibilidad a los nuevos inhibidores de la proteasa frente al VHC (fig. 1).
 genotipo 3. Adaptada de Poveda et al17.")
Principales mutaciones asociadas con resistencia a los inhibidores de la proteasa. Q80K es un polimorfismo natural que se encuentra entre el 25–39% de los genotipos 1a y está asociado con resistencia a simeprevir; D168Q se encuentra en la mayor parte de las variantes del virus de la hepatitis C (VHC) genotipo 3. Adaptada de Poveda et al17.
La resistencia a la primera generación de inhibidores de la proteasa del VHC, a la que pertenece telaprevir, se caracteriza por la selección de mutaciones en las posiciones 36, 54, 55, 155, 156, y 1779,10. El perfil de mutaciones de resistencias está influenciado por el subtipo del VHC. Mientras que los pacientes infectados con el subtipo 1a seleccionan principalmente mutaciones en las posiciones 36 y 155, los infectados con el subtipo 1b lo hacen por la selección de cambios en las posiciones 36, 54, 55, 156 y 170. Esta diferente vía para la selección de resistencias parece explicarse porque en el subtipo 1b del VHC se necesitan 2 cambios nucleotídicos en la posición 155 para producir resistencia (R155K: CGG-AAG), mientras que solamente es necesario un cambio para el subtipo 1a (R155K: AGG-AAG). Esta observación se traslada a la práctica clínica, en la que se observa una mayor barrera genética para la resistencia en el subtipo 1b que en el 1a6,7,26,27.
En general hay una amplia resistencia cruzada entre los inhibidores de la proteasa, siendo comunes a todos ellos las mutaciones en las posiciones 155 y 168. Sin embargo, como se observa en la figura 1, los cambios seleccionados en los fracasos a telaprevir no son exactamente los mismos que los observados en los fracasos a la segunda generación de inhibidores de la proteasa (p. ej., vaniprevir, simeprevir, o BI-1335)6,7,15–17. MK-5172 es un nuevo inhibidor de la proteasa en desarrollo que presenta actividad antiviral frente a variantes del VHC que presentan las mutaciones V36A/M, T54A/S, R155K/Q/T, A156S, V36M+R155K o T54S+R155K. Además, MK-5172 también ha demostrado actividad frente a todos los genotipos del VHC30.
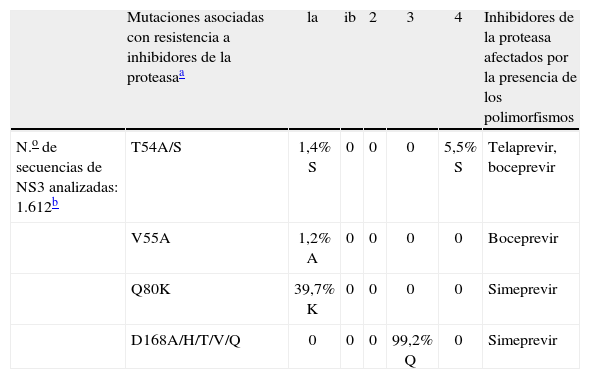
La prevalencia de polimorfismos naturales asociados con resistencias a inhibidores de la proteasa del VHC se ha evaluado en diferentes estudios en pacientes naïve. En general, y utilizando secuenciación poblacional, la prevalencia de mutaciones en las posiciones 36, 155, 156 o 168 es inferior al 1%, y entre el 3–7% para las posiciones 54 o 55. Sin embargo hay un polimorfismo, Q80K, que se encuentra entre un 25–39% de los subtipos 1a, que se ha asociado con una reducción de la sensibilidad a simeprevir de 10 veces17–19,22,23 (tabla 2). El impacto de la presencia de este polimorfismo en la respuesta virológica de simeprevir se ha evaluado recientemente en la fase II del ensayo clínico ASPIRE31. La tasa de respuesta viral sostenida (RVS) a simeprevir a dosis de 100mg fue significativamente menor en los genotipos 1a portadores de la mutación Q80K en comparación con los que no la tenían (el 22 frente al 70%, respectivamente). Sin embargo, en los pacientes infectados por el genotipo 1a que recibieron simeprevir a dosis de 150mg ya no se observaban diferencias en la RVS entre los pacientes portadores del polimorfismo y los que no lo tenían (el 61 frente al 66%, respectivamente). Hay que destacar que, independientemente de la presencia del polimorfismo Q80K, las tasas de RVS en genotipo 1a fueron menores que en genotipo 1b (el 63 frente al 80%, respectivamente), igual que se ha observado para otros inhibidores de la proteasa. Hay otro polimorfismo, D168Q, que se encuentra de forma mayoritaria en los genotipos 3 (> 99%) y que es el responsable de la resistencia natural a simeprevir en estas variantes32.
Prevalencia de polimorfismos naturales asociados con resistencia a inhibidores de la proteasa en los diferentes genotipos/subtipos del virus de la hepatitis C (VHC).
| Mutaciones asociadas con resistencia a inhibidores de la proteasaa | la | ib | 2 | 3 | 4 | Inhibidores de la proteasa afectados por la presencia de los polimorfismos | |
| N.o de secuencias de NS3 analizadas: 1.612b | T54A/S | 1,4% S | 0 | 0 | 0 | 5,5% S | Telaprevir, boceprevir |
| V55A | 1,2% A | 0 | 0 | 0 | 0 | Boceprevir | |
| Q80K | 39,7% K | 0 | 0 | 0 | 0 | Simeprevir | |
| D168A/H/T/V/Q | 0 | 0 | 0 | 99,2% Q | 0 | Simeprevir |
Adaptada de Poveda et al17.
Los ensayos clínicos en fases II y III (ADVANCE3, ILLUMINATE4 y REALIZE5) han permitido identificar las mutaciones asociadas con resistencia (RAV) que se seleccionan en pacientes que no alcanzan RVS con terapia combinada con telaprevir y Peg-IFN/RBV. En un subanálisis de estos estudios se observó que el 84% de los pacientes infectados con genotipo 1a y el 54% de los infectados con genotipo 1b que no alcanzaban RVS presentaban alguna variante resistente. Se observó cómo las mutaciones V36A/M, T54A/S, R155K/T, A156S/T y D168N se seleccionaron en los pacientes que no alcanzaron RVS, con diferencias significativas respecto a su detección en las muestras basales. Se observaron también otras RAV en los pacientes que no alcanzaron RVS, pero sin alcanzar diferencias significativas en su detección con respecto al momento basal, como fueron las variantes V36G/I, I132V (solo en el subtipo 1a, ya que este es un polimorfismo natural en el subtipo 1b), R155G/M y A156F/N/V.
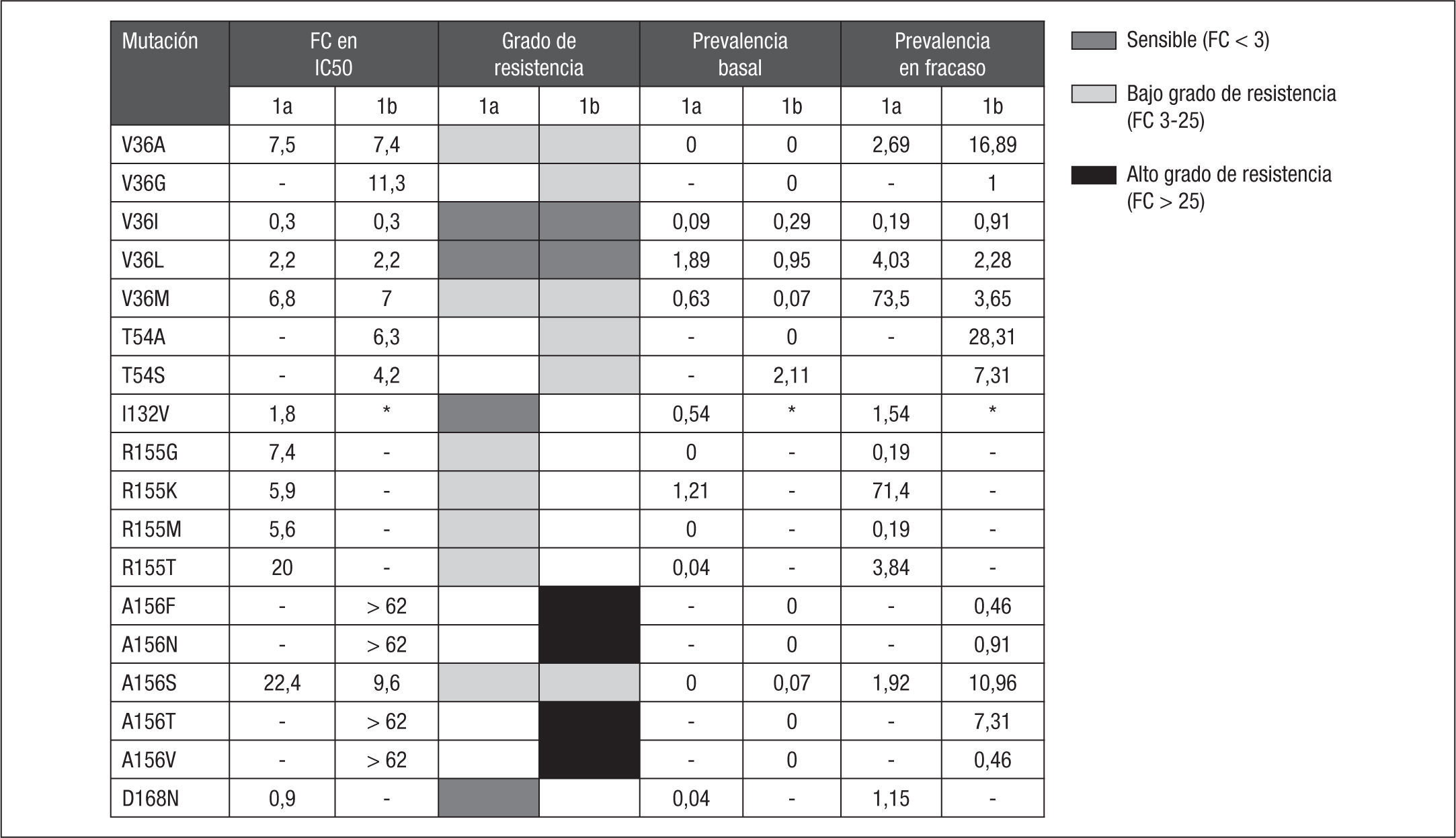
El impacto específico de cada una de estas mutaciones sobre la susceptibilidad in vitro a telaprevir puede ser cuantificado, estableciendo los cambios específicos en el fold-change (FC) para cada mutación frente a una cepa salvaje. Mediante estudios fenotípicos9,10 se ha podido establecer el FC de cada una de estas mutaciones, que oscila entre 0,3 y > 62 (fig. 2). Estableciendo como punto de corte un FC > 3 para identificar la resistencia a telaprevir, Kieffer et al10 agrupan las RAV en las que confieren bajo grado de resistencia, cuando el FC está entre 3 y 25, y las que confieren un alto grado de resistencia, cuando el FC es > 25. Siguiendo esta clasificación, las mutaciones V36A/G/M, T54A/S, R155G/K/M/T y A156S confieren bajo grado de resistencia, mientras que los cambios A156F/T/V y la combinación V36M+R155K confieren alto grado de resistencia a telaprevir. Las mutaciones V36I/L, I132V y D168N, por sí solas, no confieren resistencia a telaprevir (fig. 2). En los fracasos virológicos con telaprevir, un 36% de las RAV seleccionadas produce alto grado de resistencia (FC > 25): V36M+R155K (32%), A156S (2%) y A156T (2%).

Mutaciones de resistencia seleccionadas en pacientes que no alcanzan respuesta virológica sostenida en las fases II/III de los ensayos clínicos con telaprevir + interferón pegilado y ribavirina. *I132V es un polimorfismo natural en el subtipo 1b. Adaptada de Kieffer et al10.
En función del subtipo genético, 1a frente a 1b, se han establecido diferentes rutas de aparición de las RAV en pacientes que no alcanzan RVS en terapia combinada con telaprevir10. En los pacientes con subtipo 1a, las RAV que predominaron fueron V36M (8%), R155K (13%) y V36M+R155K (46%, alto grado de resistencia), y solo en un 14% de los casos no se detectó RAV. En los pacientes con subtipo 1b, el patrón de resistencias fue diferente: V36A (8%), T54A (18%) y A156T/V (10%, alto grado de resistencia), con hasta un 44% de pacientes en los que no se detectaron RAV.
En la mayoría de los pacientes que no alcanzan RVS a telaprevir en combinación con Peg-IFN/RBV se seleccionan mutaciones asociadas con resistencia (el 58% de los pacientes del ADVANCE y el 71% del REALIZE). La prevalencia y las características de las mutaciones de resistencia seleccionadas fueron diferentes en función de las características del paciente10. En pacientes naïve a tratamiento (ADVANCE, semana 12), un 12% de los que no alcanzaron RVS seleccionó mutaciones de resistencia, con una distribución similar entre los que seleccionaron mutaciones con alto y bajo grado de resistencias en los pacientes infectados con genotipo 1a, mientras que en los que presentaban genotipo 1b solo se detectaron mutaciones asociadas a bajo grado de resistencia. En los pacientes que previamente habían experimentado una recidiva a un tratamiento con Peg-IFN/RBV (REALIZE), solo el 6% de los que no alcanzaron RVS seleccionó mutaciones de resistencia, y la mayoría lo hizo con RAV asociadas con baja resistencia. Sin embargo, entre los pacientes no respondedores o respondedores parciales a terapias previas con Peg-IFN/RBV (REALIZE), hasta el 40% de los pacientes seleccionó mutaciones de resistencia con una proporción elevada de RAV, que confieren alto grado de resistencia. Como era de esperar, en pacientes en los que se interrumpe el tratamiento de forma precoz (menos de 4 semanas), por causas diferentes al fracaso virológico, mayoritariamente se observan variantes virales sin mutaciones de resistencia.
Finalmente destacar que en un subestudio del ensayo REALIZE se demostró que la selección de RAV no se ve afectada por la realización o no de la fase “lead in”. Las tasas globales de selección de RAV fueron del 22 frente al 21%, respectivamente.
Impacto de la presencia de mutaciones de resistencia de forma basal en la respuesta a telaprevirCon respecto a la detección de RAV basales se han presentado hasta el momento datos de diferentes estudios que han evaluado la relación entre su presencia y la probabilidad de alcanzar RVS. Mediante el empleo de técnicas de secuenciación poblacional se ha demostrado que los cambios en las posiciones asociadas con resistencia a telaprevir son poco frecuentes en pacientes que no han sido expuestos al fármaco11,18,19 y que, además, no existe una asociación clara entre su presencia y la posibilidad de no alcanzar RVS18,33,34, aunque, como se verá más adelante, puede haber alguna consideración especial para los pacientes no respondedores a terapias previas con Peg-IFN/RBV.
Kuntzen et al19 evaluaron la prevalencia de las diferentes RAV en la proteasa del VHC de 507 pacientes naïve infectados por subtipo 1a (n=362) y 1b (n=145). En los pacientes con subtipo 1a se detectaron los cambios R155K (0,8%), V36L/M (1,7%) y T54S (1,7%); entre los pacientes infectados con subtipo 1b, solo se detectó el cambio T54S (1,4%). Kieffer et al10, en el estudio más amplio que se ha presentado, muestran los datos de 3.449 secuencias basales de proteasa de todos los pacientes incluidos en los ensayos clínicos de telaprevir, detectando variantes asociadas a resistencia (secuenciación poblacional) en el 5% de los pacientes. La mayoría de estos pacientes presentaron la mutación V36L (1,5%) o T54S (2,7%), en ambos casos se trata de mutaciones con escaso impacto fenotípico sobre la resistencia a telaprevir. La prevalencia en el caso de mutaciones con mayor impacto en la resistencia fue muy baja (< 0,6%): V36M (0,3%), T54A (0,03%) y R155K (0,5%). En cambio I132V se detectó con una prevalencia del 0,5% (subtipos 1a).
Cuando se evalúa el impacto que la detección de RAV basales puede tener en la respuesta a la terapia triple con telaprevir, se observan, en general, cifras similares de RVS en los pacientes que inician tratamiento y presentan variantes de resistencia y los que inician con virus no portadores de estas variantes (el 70 frente al 72%, respectivamente). Sin embargo, en los pacientes no respondedores a un tratamiento previo con Peg-IFN/RBV, solo el 14% de los que presentaron mutaciones de resistencia en la muestra basal alcanzó RVS, a diferencia de los que no tenían mutaciones basales, en los que se alcanzó RVS en el 33%. Además, en este tipo de pacientes se ha comprobado que los que presentaban las variantes V36M y R155K no alcanzaron RVS, mientras que sí se alcanzó RVS en la mayoría de los pacientes naïve a tratamiento que presentaron estas variantes. Parece, por tanto, que la presencia de RAV basales puede limitar la posibilidad de alcanzar RVS en determinadas poblaciones, como la de los pacientes no respondedores a Peg-IFN/RBV10.
De Meyer et al35 presentan los datos del subestudio virológico del REALIZE con datos similares. El estudio se realizó en un total de 662 pacientes, de los que finalmente pudieron analizar la proteasa de 652 pacientes (98%). En estos, solo el 3% presentó variantes asociadas a resistencia (n=18), el 1,8% (n=12) la mutación T54S, el 0,6% (n=4) R155K y el 0,3% (n=2) V36M. Como era de esperar, al igual que en el estudio de Kieffer et al10, que incluía también a estos pacientes, son los pacientes que no han respondido a terapias previas con Peg-IFN/ RBV en los que se observa un efecto de las RAV basales sobre la probabilidad de alcanzar RVS, ya que ningún no respondedor previo a Peg-IFN/RBV con RAV basales (n=5) alcanzó RVS.
Hay que destacar que todos estos estudios, a excepción de los datos presentados del estudio EXTEND36, se han realizado utilizando secuenciación poblacional. La tecnología utilizada en el EXTEND permite investigar RAV con mayor sensibilidad a la secuenciación poblacional, con un límite de sensibilidad del 5%. Hasta el momento se han presentado algunos estudios preliminares utilizando tecnologías más sensibles (MAMA-PCR37, Ultra Deep Sequencing38–40), que permiten detectar RAV con una sensibilidad ≤ 1%. Los resultados de estos estudios, que incluyen, en general, un escaso número de pacientes, deben ser interpretados hasta el momento con precaución.
En el momento actual, el estudio de mutaciones de resistencias basales antes del inicio de una terapia con telaprevir en combinación con Peg-IFN/RBV, tiene un carácter exclusivamente académico y de investigación, y no parece que haya hasta el momento datos que justifiquen su aplicación en la rutina asistencial en este contexto en particular.
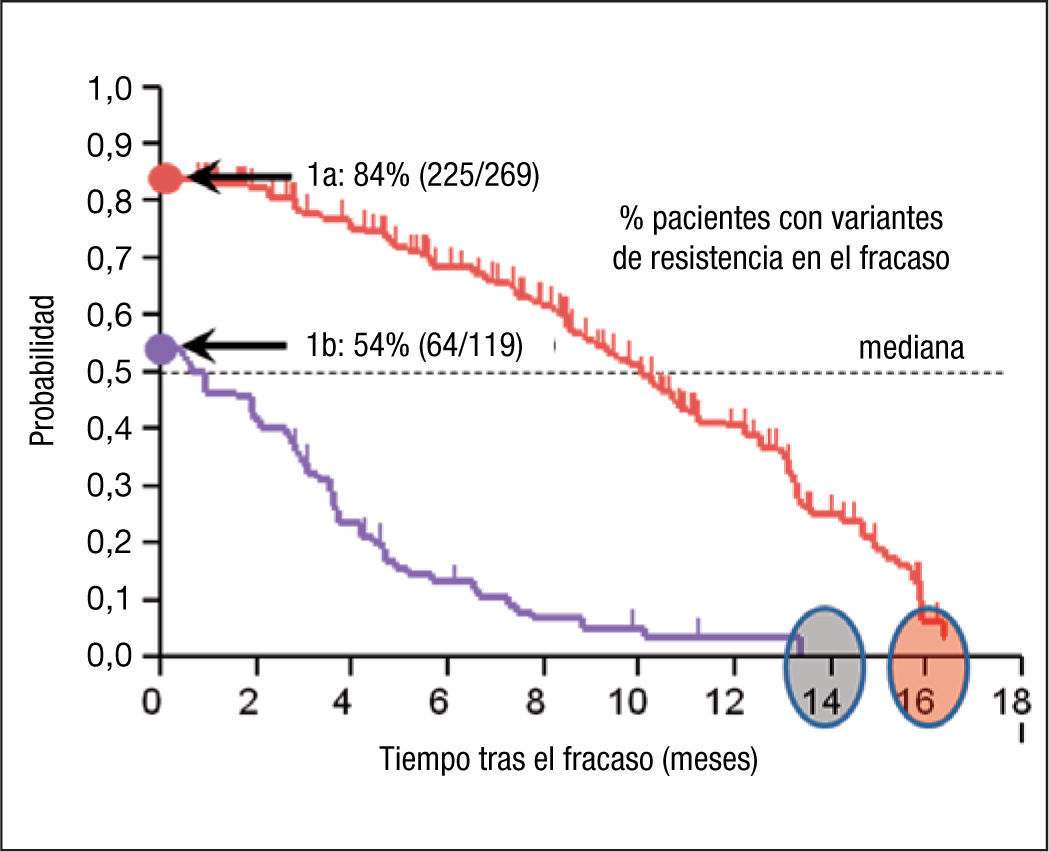
Dinámica de reversión de las variantes de resistencia a telaprevirUna vez seleccionadas, las variantes de resistencia a telaprevir, en ausencia de presión farmacológica, tienden a revertir en el tiempo. El 77% de los pacientes que no alcanzaron RVS a telaprevir en combinación con Peg-IFN/RBV en los ensayos en fase III, presentaron alguna variante de resistencia (254/388). En el 60% de estos pacientes (153/254), las variantes de resistencia dejaban de detectarse en una mediana de 9,7 meses mediante métodos de secuenciación poblacional. No se han encontrado diferencias en la tasa de desaparición de la RAV en función de que los pacientes fueran naïve o pretratados. Sin embargo, la mediana de tiempo a la reversión de cada mutación es diferente41, hecho que puede explicarse por el diferente impacto de estas sobre la capacidad de replicación del VHC9,10,42–44. Además, también se han descrito diferencias en la dinámica de reversión de las variantes en los genotipos 1a y 1b, constatándose un efecto dependiente del subtipo en la reversión, que se produce más rápidamente en los pacientes con genotipo 1b (mediana 0,9 meses [intervalo de confianza —IC— del 95%, 0,00–2,10], estimación Kaplan Meier, n=269] que en los 1a [mediana 10,7 meses [IC del 95%, 9,6–12,4], estimación Kaplan Meier, n=119] (fig. 3). De Meyer et al35 han realizado una descripción detallada de la dinámica de reversión en los pacientes del estudio REALIZE.
 transcurrido hasta la reversión de mutaciones de resistencia seleccionadas en los fracasos a telaprevir en los genotipos 1a y 1b del virus de la hepatitis C (VHC) una vez suspendido el tratamiento. Adaptado de Sullivan et al41.")
Estimación de Kaplan-Meier del tiempo (meses) transcurrido hasta la reversión de mutaciones de resistencia seleccionadas en los fracasos a telaprevir en los genotipos 1a y 1b del virus de la hepatitis C (VHC) una vez suspendido el tratamiento. Adaptado de Sullivan et al41.
Hay menos datos en relación con el uso de tecnologías que permitan una mayor sensibilidad en la detección de RAV que la secuenciación poblacional. Se han presentado datos preliminares del estudio EXTEND36, en el que se ha realizado un análisis clonal en las muestras basales, con una sensibilidad del 5%, verificándose cómo en este caso las RAV desaparecen en una mediana de 22 meses tras la suspensión del tratamiento. En esta misma línea, y también utilizando análisis clonal, se encuentran los datos presentados por Susser et al45, con una mediana de seguimiento de 4,2 años.
La reversión de las RAV tiene una implicación clínica importante, la posibilidad de la utilización de estos fármacos en futuros esquemas de tratamiento, previa confirmación mediante un estudio de resistencias en el que se confirme la ausencia de estas mutaciones. No obstante, en el momento actual, se hacen necesarios más estudios que validen estas estrategias de “reciclaje” de fármacos.
AgradecimientosFondo de Investigación Sanitaria (CP08/00214, PI10/02166) y Red Temática Cooperativa de Investigación en SIDA del FIS (ISCIII-RETIC006/ 0016).
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

