





New massive sequencing techniques make it possible to determine the composition of airway microbiota in patients with cystic fibrosis (CF). However, the relationship between the composition of lung microbiome and the clinical status of paediatric patients is still not fully understood.
Material and methodsA cross-sectional observational study was conducted on induced sputum samples from children with CF and known mutation in the CFTR gene. The bacterial sequences of the 16SrRNA gene were analyzed and their association with various clinical variables studied.
ResultsAnalysis of the 13 samples obtained showed a core microbiome made up of Staphylococcus spp., Streptococcus spp., Rothia spp., Gemella spp. and Granulicatella spp., with a small number of Pseudomonas spp. The cluster of patients with less biodiversity were found to exhibit a greater number of sequences of Staphylococcus spp., mainly Staphylococcus aureus (p 0.009) and a greater degree of lung damage.
ConclusionAn airway microbiome with greater biodiversity may be an indicator of less pronounced disease progression, in which case new therapeutic interventions that prevent reduction in non-pathogenic species of the airway microbiota should be studied.
Las nuevas técnicas de secuenciación masiva permiten determinar la composición de la microbiota de las vías respiratorias en pacientes con fibrosis quística (FQ). Sin embargo, la relación entre la composición de la microbiota pulmonar y el estado clínico de los pacientes pediátricos todavía no se ha establecido bien.
Material y métodosSe realizó un estudio transversal observacional en muestras de esputo inducido de niños con FQ y mutación conocida en el gen CFTR. Se analizaron las secuencias bacterianas del gen 16SrRNA y se estudió su asociación con diversas variables clínicas.
ResultadosEl análisis de las 13 muestras obtenidas mostró un microbioma central compuesto por Staphylococcus spp., Streptococcus spp., Rothia spp., Gemella spp. y Granulicatella spp., con un pequeño número de Pseudomonas spp. Se descubrió que el grupo de pacientes con menos biodiversidad mostraba un mayor número de secuencias de Staphylococcus spp., principalmente Staphylococcus aureus (p 0,009) y un mayor daño de la función pulmonar.
ConclusiónLa mayor biodiversidad del microbioma de vía respiratoria puede ser un indicador de menor progresión de la enfermedad, en cuyo caso deben estudiarse nuevas intervenciones terapéuticas que prevengan la disminución de especies no patógenas.
Bronchopulmonary infection is the main prognostic determinant in patients with cystic fibrosis (CF). Traditional studies, using selective culture of respiratory samples, only identify a fraction of bacterial species involved at the stable stage and at the exacerbation of the disease.1 New massive sequencing techniques make possible to determine the composition of the microbiota in CF patients which includes a large number of species in addition to the pathogens traditionally considered responsible for respiratory infections (Pseudomonas aeruginosa, Haemophilus influenzae and Staphylococcus aureus).2 Changes have been described in the microbiome of the lower respiratory tract during the different stages of the disease, but interpretation of the significance of these changes is controversial, especially in the paediatric population.3,4 The objective of this study was to correlate the clinical situation of a group of paediatric CF patients with the composition of their lung microbiota determined by massive sequencing techniques.
Material and methodsA cross-sectional observational study was performed on induced sputum samples from 13 children between the ages of 6 and 17 years with clinically stable CF and known mutation in the cftr gene. Patients were recruited at the CF Unit of the Hospital Doce de Octubre in Madrid, a centre accredited for CF assistance. This study was approved by the Ethics Committee and informed written consent was obtained from parents or legal guardian of all subjects.
During a scheduled outpatient visit a series of variables were recorded (age, gender, cftr mutation, extrapulmonary and pulmonary involvement, history of bacterial colonization, and treatments received). A sample of sputum induced by 7% hypertonic saline solution was obtained and frozen at −20°C for subsequent processing. Nucleic acids were extracted and purified with the commercial system MolYsis™ (Molzym, Germany), which allows the selective lysis of human cells with their DNA in order to isolate bacterial DNA. Parallel cultures could not be performed, due to paucity of sample volume obtained in the majority of younger patients. Historical microbiology records of cultures in the last 6 months were used to determine the infection status with classical pathogens (chronic or intermittent) of patients. Massive sequencing was performed (two strands) with the Illumina MiSeq system (Illumina, USA) and sequences of 16S (V3 and V4) were obtained using the ssu-align and meta-rna programmes.
The sequences were quality-filtered using the Pyronoise® programme and samples were compared with each other using the Unifrac programme. The bacterial taxonomy of each sample was assigned and we determined the absolute and relative abundance of species as well as the dominance and composition of bacterial communities in the samples obtained. In addition, the diversity of each sample (alpha diversity) was studied using the Shannon–Wiener index and the diversity among samples (beta diversity) using the Bray–Curtis distance and principal component analysis. Only sequences measuring between 250bp and 500bp with an end-trimming quality greater than 25 were analyzed and included in this study. The Operational Taxonomic Units (OTUs) were assigned using the RDP database (Ribosomal Database Project) for the 16S rRNA gene. All the statistical analyses were done using the statistical software SPSS version 22.0 for Windows (SPSS Inc., 2013). The results were considered statistically significant when p<0.05.
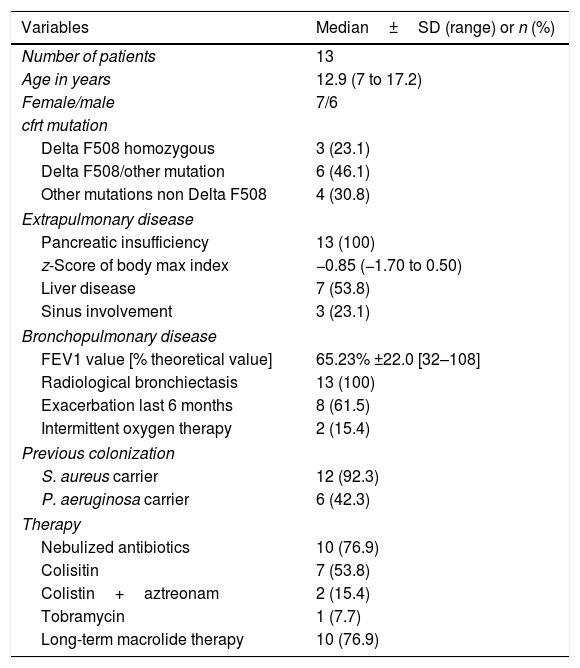
ResultsAll patients included had mutations associated with severe forms of CF and pancreatic insufficiency. Four out of the 13 patients (30.7%) exhibited severe respiratory involvement according to percentage of predicted forced expiratory volume in 1s (FEV1%). According to current microbiological criteria,5 92% of patients were chronic carriers of S. aureus and 43% chronic carriers of P. aeruginosa. None of the patients had a history of non-tuberculous mycobacteria infection in previous cultures. The main characteristics of the study subjects, variables associated with the disease, comorbidity and treatment received are shown in Table 1.
Demographic characteristics of patients and clinical variables of the disease.
| Variables | Median±SD (range) or n (%) |
|---|---|
| Number of patients | 13 |
| Age in years | 12.9 (7 to 17.2) |
| Female/male | 7/6 |
| cfrt mutation | |
| Delta F508 homozygous | 3 (23.1) |
| Delta F508/other mutation | 6 (46.1) |
| Other mutations non Delta F508 | 4 (30.8) |
| Extrapulmonary disease | |
| Pancreatic insufficiency | 13 (100) |
| z-Score of body max index | −0.85 (−1.70 to 0.50) |
| Liver disease | 7 (53.8) |
| Sinus involvement | 3 (23.1) |
| Bronchopulmonary disease | |
| FEV1 value [% theoretical value] | 65.23% ±22.0 [32–108] |
| Radiological bronchiectasis | 13 (100) |
| Exacerbation last 6 months | 8 (61.5) |
| Intermittent oxygen therapy | 2 (15.4) |
| Previous colonization | |
| S. aureus carrier | 12 (92.3) |
| P. aeruginosa carrier | 6 (42.3) |
| Therapy | |
| Nebulized antibiotics | 10 (76.9) |
| Colisitin | 7 (53.8) |
| Colistin+aztreonam | 2 (15.4) |
| Tobramycin | 1 (7.7) |
| Long-term macrolide therapy | 10 (76.9) |
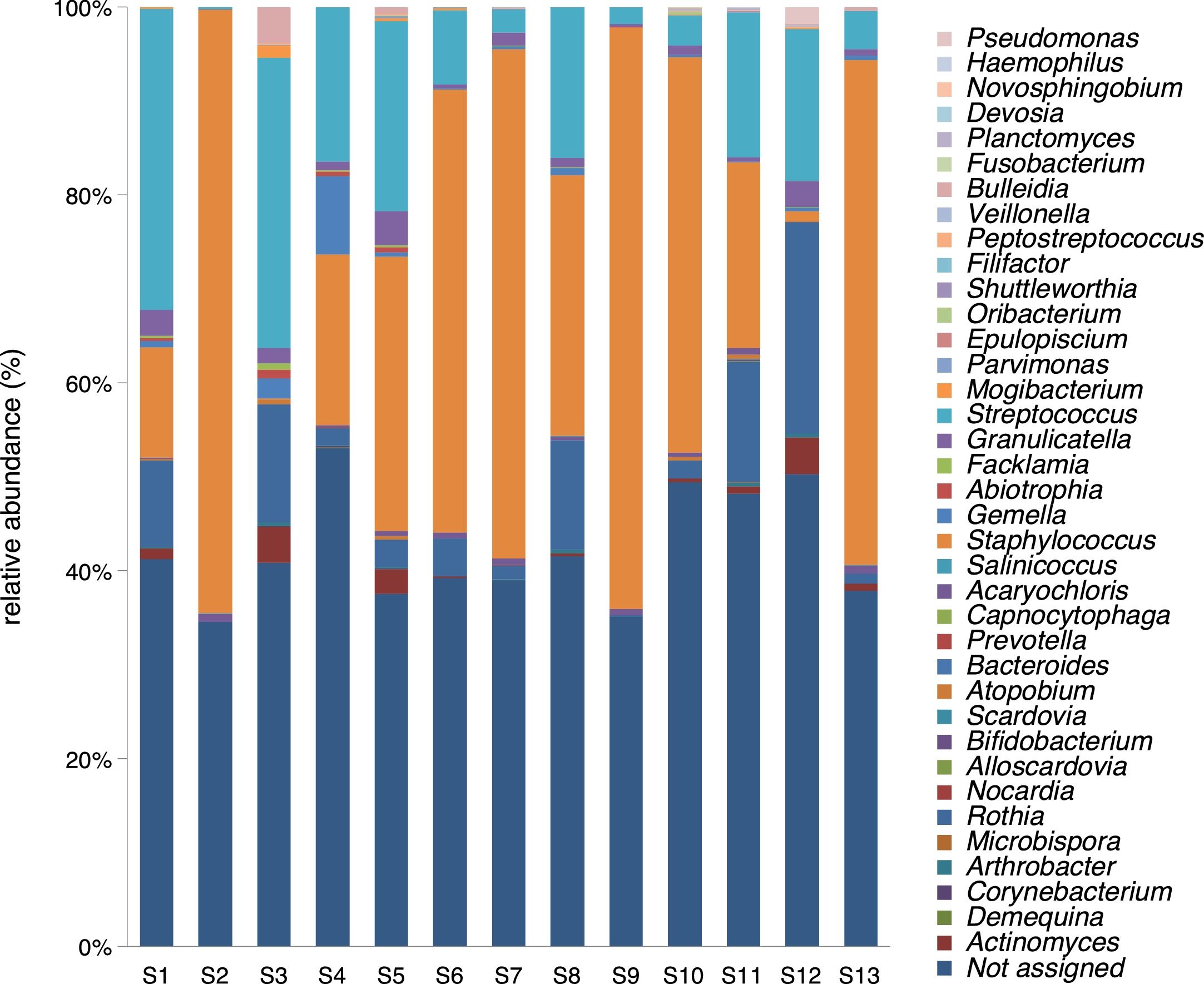
A mean of 4048 sequences were obtained in each sample, 2332 (57.6%) of which were assigned. The number of genera in the 13 samples ranged between 9 and 26, with a mean of 14.92 (standard deviation of 4.80). Five genera were found in all the samples: Staphylococcus spp., Streptococcus spp., Rothia spp., Granulicatella spp., and Gemella spp. With the exception of the latter species, the relative abundance was >1% according to the criteria of phylogenetic composition (core microbiome), defined by Coburn et al.6 (Table 2).
Comparison between the genera constituting the microbiome core.
| Genus | Relative abundance >1%a | Most abundant genusb | Dominant genusc | Maximun abundance relatived |
|---|---|---|---|---|
| Staphylococcus | 12 | 10 | 6 | 64.3 |
| Streptococcus | 12 | 2 | 2 | 32.02 |
| Rothia | 11 | 1 | 0 | 22.5 |
| Granulicatella | 6 | 0 | 0 | 3.59 |
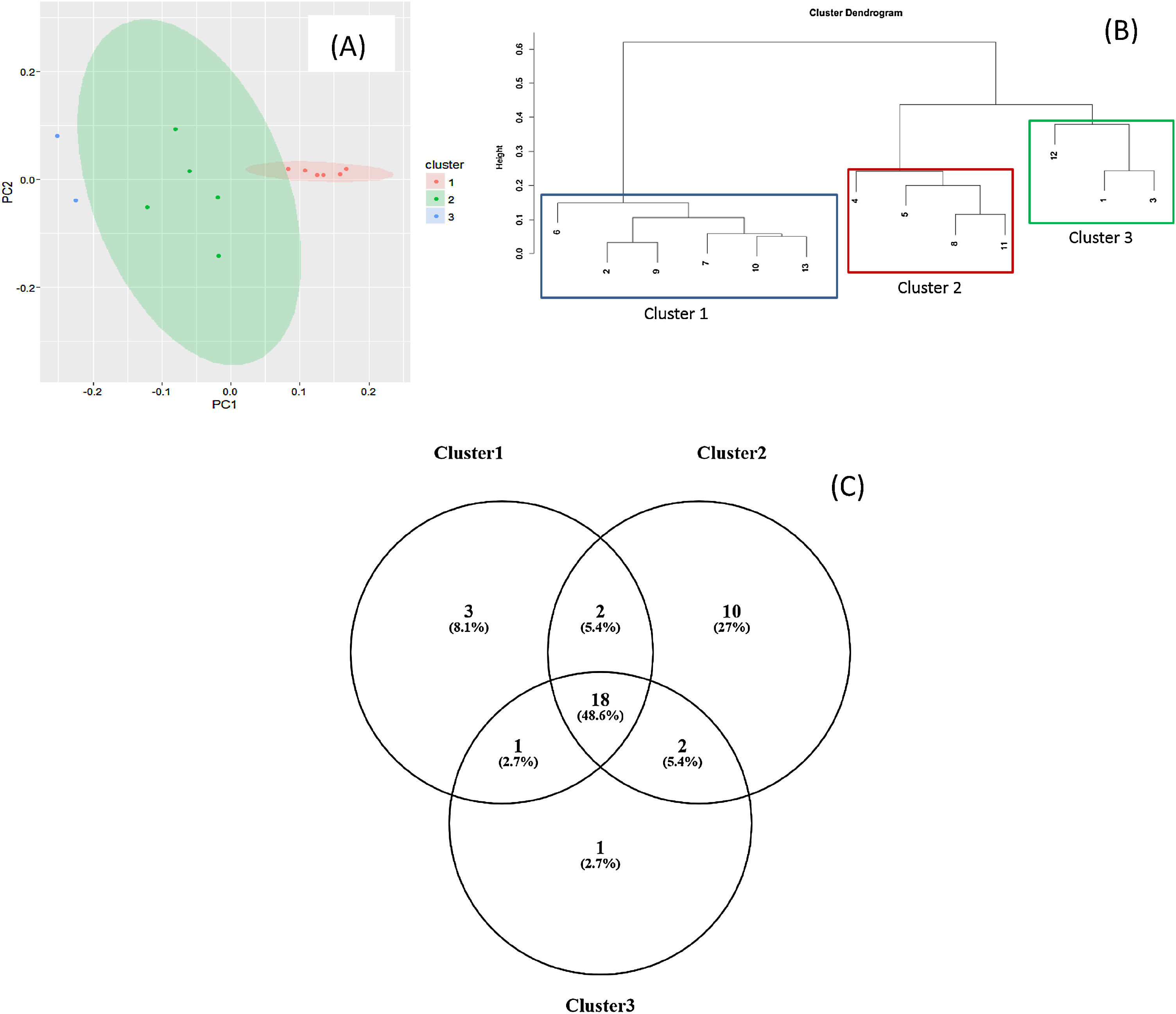
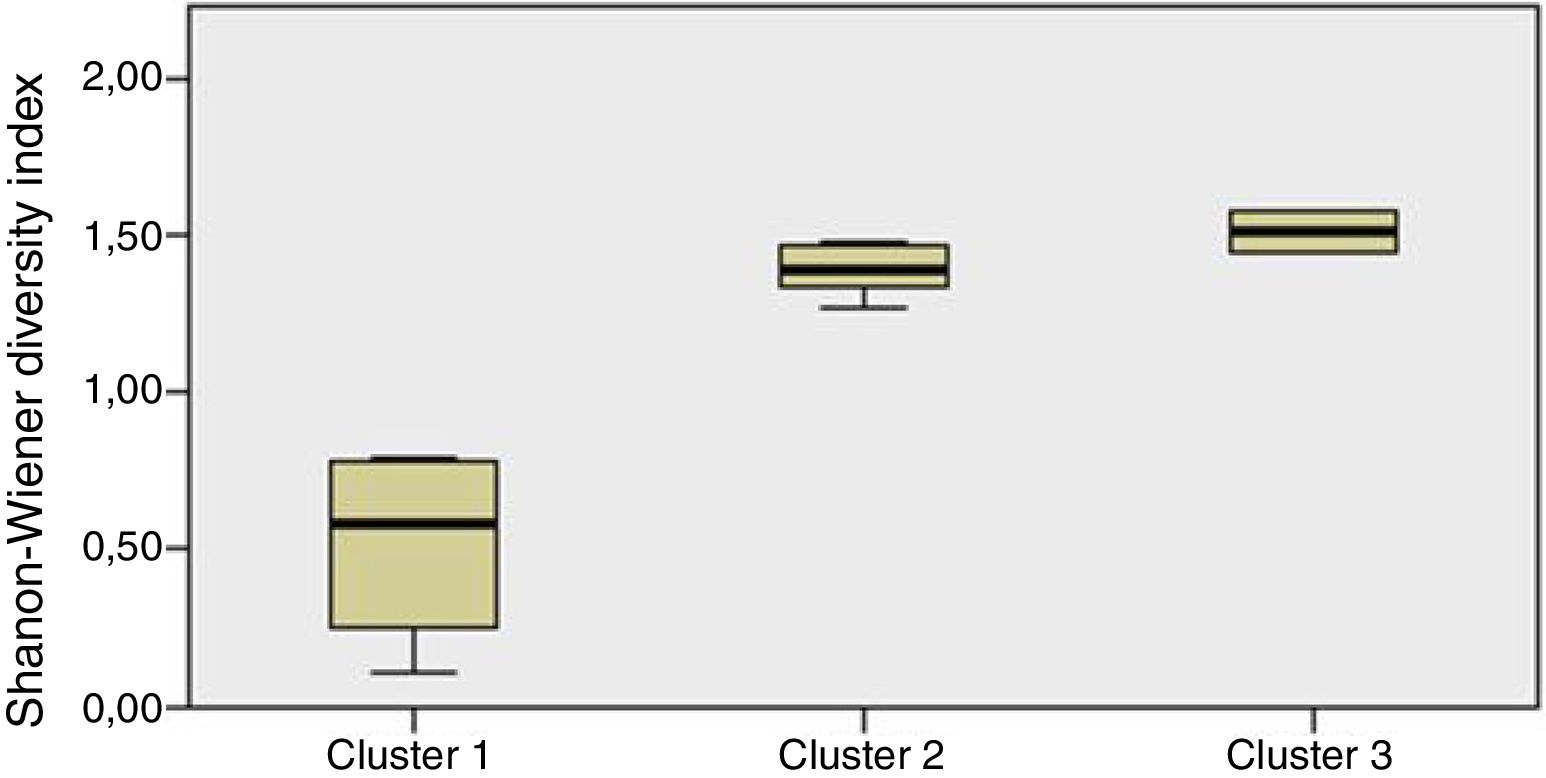
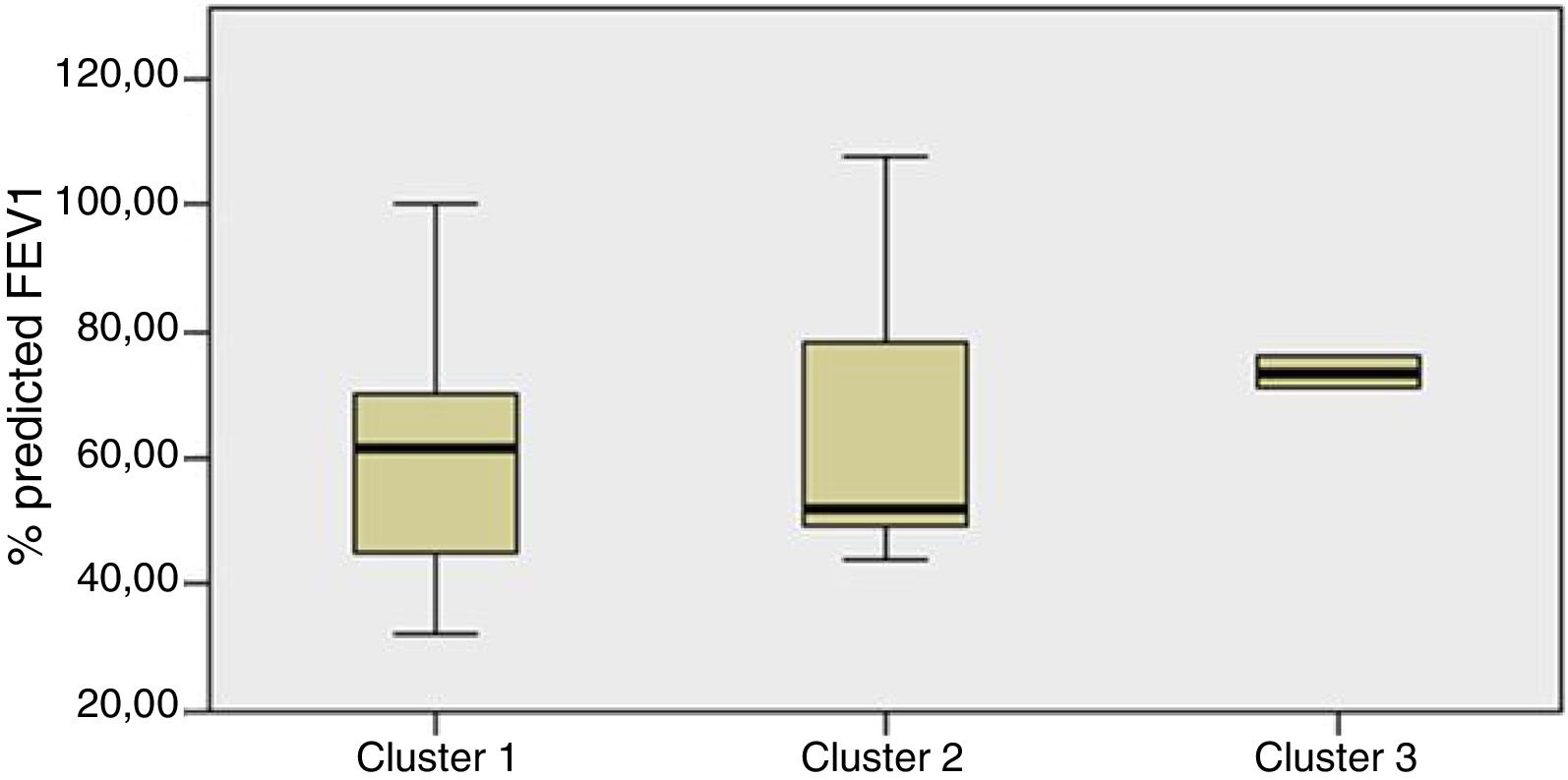
Staphylococcus spp. was the most abundant genus in 10 patients (76.92% of the total) and dominant5 in 6 (46.15% of the total), whereas Streptococcus spp. was the most abundant and dominant species in 2 patients (15.38% of the total) (Fig. 1). Haemophilus spp. was detected in the microbiome of 3 patients (23.07% of the total). Staphylococcus spp. was present in all patients, with S. aureus being the most frequent species in all the samples, followed by Staphylococcus epidermidis. Pseudomonas spp. was detected only in 2 patients (15.38%), and in both cases accounted for less than 5% of the total number of genera. Fourteen genera only appeared in a maximum of one sample. Stenotrophomonas spp., Achromobacter spp., Mycobacterium spp. and Burkholderia spp. were not detected in any of the samples. The alpha diversity values, expressed as Shannon–Wiener indexes, ranged from 0.11 to 1.57, with a mean value of 1.00. When the 13 samples were compared using a principal component analysis (PCA) 3 clusters were found (Fig. 2a–c). Patients in group 1 exhibited the lowest alpha diversity, whereas those in group 3 exhibited the highest, and these differences were statistically significant (p=0.009) (Fig. 3). In addition to the greater number of species, there was a greater density of communities of Staphylococcus spp., a smaller number of colonies of Streptococcus spp. and Actinomyces spp. and a smaller absolute number of genera in group 1, although the differences were not statistically significant. No statistically significant associations were found between clinical variables and different diversity indexes. However, it was observed a trend between cluster defined by PCA and degree of respiratory involvement, defined by theoretical % FEV1 (Fig. 4). Cluster 1, with the lowest alpha diversity, expressed by Shannon index, showed the lowest FEV1 values.

 Principal component analysis (PCA) for the microbiome composition of the samples; (b) dendrogram; (c) Venn diagram.")
.")
 and clusters determined by PCA for the microbiome composition. Kruskal–Wallis test for indenting samples (p=0.577).")
New massive sequencing techniques provide a great amount of additional information in patients with CF. For example, certain bacterial species, whose clinical significance is not clear, may be detected by these new techniques but not by traditional procedures.7 Advances in molecular microbiology have shown that lung infection in CF is polymicrobial8 and the pathogen abundance and type vary depending on the individual and on the disease stage.9
As described in other studies,6,10,11Staphylococcus spp., Streptococcus spp. and other genera such as Granulicatella spp., Gemella spp., Rothia spp., and Actinomyces spp. were regularly detected in the respiratory tract of CF patients, and it was also observed a certain degree of variability among individuals in the structure, composition and diversity of the bacterial communities. By PCA analysis it was possible to establish clearly three distinct groups. These differences were found even though the group was homogeneous, delimited within an age group and with no apparent association with mutation type or other variables such as age or treatment administered.
Clinical records showed that less than a half (38%) of our patients were chronic carriers of P. aeruginosa as determined by the traditional culture, while the great majority were carriers of S. aureus, which coincides with the findings of other studies performed on this age group.12 It is remarkable that the genus Pseudomonas spp. was detected in the microbiome of only two patients of our sample. In another study performed with bronchoalveolar lavage samples from children with CF, it was reported that P. aeruginosa was also detected in the culture but not in the molecular analysis of the microbiota.13 This apparent discrepancy may be due to the magnification of Gram-negative bacilli usually found with traditional culture methods.14 As these methods are intentionally selective, they are unable to detect the vast majority of the microbioma components. The amount of P. aeruginosa in the airway microbiota progressively increases, especially at the beginning of adulthood and tends to predominate over other species, with a resulting decrease in the amount of Staphylococcus spp., due to the antagonism between the two genera.15 The existence of this inverse relationship is supported by the fact that prophylactic treatment to prevent colonization by S. aureus may lead to an increase in the incidence and prevalence of infection due to P. aeruginosa.16
When the microbiome in the different samples was compared using a principal components analysis (PCA), there was a clearly different group with a higher density of Staphylococcus spp. as opposed to the other two groups in which there was a higher density of Actinomyces spp. and Streptococcus spp. There was a smaller absolute number of genera and lower alpha diversity expressed as a Shannon index in the group with the higher density of Staphylococcus spp. This inversely proportional relationship between Staphylococcus spp. and alpha diversity has been reported in other studies.17 On other hand, the predominance of Streptococcus spp. is associated with a microbiome of greater diversity,18 which usually occurs in patients in whom there is less progression of lung disease. In our study, a microbiome with low alpha diversity, dominated by Staphylococcus spp. and with less number of genera found in non-CF children seems to be in relationship with lower values of FEV1, but without statistically significant differences.
An association between deteriorated lung function and alpha diversity has been described in cross-sectional studies.19 The diversity of bacterial communities in healthy individuals is greater than that in CF patients.20 This loss of diversity in CF occurs progressively with age and lung disease.21 The microbiome of infants and toddlers without lung damage appears to be dominated by Streptococcus spp. and anaerobic genera such as Prevotella spp. and Veionella spp.,22 as in children without CF. Despite the lack of longitudinal studies, diversity seems to increase during the first decade of life, a period when lung function is usually relatively conserved.6 The diversity begins to decrease from adolescence to adulthood in parallel with lung function parameters and airway remodelling due to progression of the disease.
New microbiological methods reveal a high number of anaerobic microorganisms in the airways of CF patients.23 The role played by these organisms is not clear, and it is not known whether they are commensal organisms with a certain protective effect against traditional pathogens. In our study, as occurs in other studies of patients with CF24 and in patients with persistent lung infections such as chronic obstructive pulmonary disease,14 a small number of Streptococcus spp. and Actinomyces spp. seems to be in relationship with a worsening of respiratory parameters. In our study, the genus Mycobacterium spp. was not detected. This may be due to the fact that non-tuberculous mycobacteria are mainly found in long term CF patients, over 15 years old,25,26 whereas in our sample the majority of patients were younger individuals. Some authors consider that sputum samples are not suitable for studying the lower airway microbiota.27 However, other studies have shown that the contact of the sample with oral bacteria has a discrete relevance in lower airway microbiota composition analysis.28
Implementation of the new methodologies suggest that disruption of the ecological balance of microbial communities in the lungs, loss of mutualism, and the emergence of competing species may be determining factors in a variety of respiratory diseases such as CF.29,30 More studies must be performed in order to determine whether changes observed in the microbiome of patients with CF play a causal role in the evolution of pulmonary disease, or are only a consequence of the progression of the disease and the antibiotic treatments received. In first premise, the studies must offer data that allow interventions on airway microbiome with positive repercussion in the clinical evolution of CF.
FundingThis work has been funded with a grant from the foundation Navarro Tripodi.
Conflicts of interestNothing to declare.
To Dr. Gloria García Hernández, for a lifetime of dedication to the care of children with cystic fibrosis.
