


Las enfermedades crónico-degenerativas son actualmente susceptibles de ser estudiadas a través de sus alteraciones genómicas estructurales y funcionales. Las enfermedades neoplásicas dentro de éstas, son el grupo más estudiado en los contextos subcelular/genómico/epigenómico y proteómico.
El progreso de las tecnologías de segunda y tercera generación de secuenciación en el estudio genómico ha mejorado en la resolución de las determinaciones y en el análisis masivo simultáneo de los procesos celulares fenotípicos. Muchas de estas características moleculares sirven de indicadores o de biomarcadores fisiopatológicos de las células tumorales. Cada tipo de tumor es transformado particularmente por cambios moleculares cuasi-específicos. Algunos de los biomarcadores moleculares de tipos específicos tumorales, han sido validados como indicadores de comportamiento biológico, constituyendo factores pronósticos o predictivos clínicos, el ejemplo más significativo ha sido el uso de un microarreglo simplificado de expresión génica de 70 genes (MammaPrint), con utilidad pronóstica en pacientes con cáncer de mama en etapa clínica inicial. Asimismo, la identificación de algunos de estos biomarcadores ha favorecido el éxito de tratamientos molecularmente dirigidos, como el imatinib, en pacientes con leucemia mielocítica crónica y con sarcomas estromales de tubo digestivo.
En la última sección de esta revisión, se analiza el cáncer esporádico de mama empleando taxonomía molecular de acuerdo a sus alteraciones genómicas integrales, y comentamos su utilidad clínica.
Recently, chronic-degenerative diseases can be studied though a structural and functional genome point of view. Among these diseases, cancer is the main group studied in a subcelular/genomic/epigenomic and proteomic context.
The second and third generation of sequencing technologies used in genomic studies have improved the resolution and in massive whole-gene simultaneous analysis of the phenotypical cell processes. Many of these molecular characteristics are used as physiopathologic indicators or biomarkers of phenotype cell processes. Each tumor is particularly transformed by cuasi-specific molecular changes. Some of the biomarkers of specific tumors, have been validated as biological behavioral indicators, which are associated to prognostic or predictive clinical factors, the most significant example of these biomarker is the MammaPrint, a simplified gene expression array used with prognostic utility in early breast cancer patients. The identification of some biomarkers has favoured the targeted drugs success, such as imatinib in CML and in GISTs patients.
In the last section of this minireview, we analyze the sporadic breast cancer, through molecular taxonomy in accordance with their comprehensive genomic alterations, and we discuss their clinical utility.
Introducción
El mayor grupo de enfermedades neoplásicas que se presentan en los seres humanos corresponden a cánceres esporádicos; los cuales se desarrollan a partir de la acumulación de alteraciones moleculares en el genoma de las células somáticas. En los últimos 30 años, los progresos en las tecnologías y análisis de la secuenciación del genoma humano y particularmente del genoma de las células cancerosas han permitido construir mapas genómicos taxonómicos de los principales tipos y subtipos de tumores malignos. La identificación de las alteraciones genómicas estructurales junto con la determinación de sus perfiles de expresión genómica ha favorecido la identificación de biomarcadores pronósticos y ha ayudado a direccionar la construcción de medicamentos anticancerosos específicos, empleando como blancos terapéuticos a moléculas bioquímicamente alteradas.
Más de 400 genes han sido identificados como oncogenes (OG) y genes supresores tumorales (TSG) en el proceso de iniciación y progresión tumoral. Los OG y TSG adquieren alteraciones genómicas estructurales y funcionales, las cuales favorecen el proceso de transformación celular. Las principales alteraciones corresponden a mutaciones que aumentan el perfil transcripcional (OG), o mutaciones que bloquean la codificación funcional de la proteína (TSG). Las mutaciones en los OG afectan generalmente a un alelo (afectación heterocigota) y las mutaciones en los TSG afectan generalmente a los 2 alelos (provocando pérdida de la heterocigosidad). La mayoría de las mutaciones somáticas en las células cancerosas corresponden a sustituciones nucleotídicas, insersiones y deleciones pequeñas (indels), alteraciones en el número de copias de regiones particulares del DNA (CNVs) de alrededor de 1 Kbp de extensión, o de regiones mayores de varias Kbp a escasas Mbp, y de rearreglos cromosómicos1.
La identificación de los distintos genomas en las células tumorales ha tenido como antecedente directo el estudio progresivo de la secuenciación del genoma humano, obtenido a través de los proyectos Genoma Humano, HapMap y 1000 Genomas del DNA de algunos tipos de células somáticas normales (por ejemplo, leucocitos, células epiteliales) diferenciadas a partir de células germinales, el que frecuentemente es denominado germline-DNA. En la última década, además de los estudios genómicos estructurales, se ha obtenido el perfil transcriptómico tanto del germline-DNA como del genoma de los principales tipos y subtipos de cáncer1. Recientemente se ha empezado a estudiar la participación de las regiones no-codificantes del DNA en la regulación de la expresión génica2.
En este artículo se analizan las principales alteraciones genómicas estructurales y funcionales que son adquiridas por las células neoplásicas en los cánceres esporádicos, remarcando la participación de las alteraciones en el número de copias de regiones particulares del DNA; y asimismo se examina el cáncer mamario como modelo de dichas alteraciones.
Alteraciones genómicas estructurales y funcionales adquiridas por las células tumorales en el desarrollo de cánceres esporádicos
El modelo fisiopatológico subcelular/epi/genómico/proteómico más estudiado en el contexto de la Biología Celular y Molecular es el cáncer. Los fenotipos particulares de las poblaciones celulares del tumor primario y del contexto global de respuesta del hospedero (paciente), interactúan estrechamente para producir una relación huésped/tumor o comportamiento biológico particular en la dinámica de la enfermedad (horizonte clínico). Se han reconocido más de 200 tipos de neoplasias malignas (y aproximadamente 10 subtipos de cada uno), de acuerdo a su localización topográfica y al tipo de célula de origen (fenotipo morfológico). Cada tipo de cáncer cursa con alteraciones funcionales globales parecidas en su transformación, sin embargo un mismo tipo e incluso diferentes subtipos, cursan con alteraciones funcionales y estructurales particularmente específicas (fenotipo funcional)3, que sumadas a las características fenotípicas del individuo relacionadas con el mantenimiento y preservación de su homeostasis4, conducen a interacciones biológicas específicas que traducen finalmente el horizonte clínico de la evolución del padecimiento. Las características fenotípicas sistémicas del organismo humano para preservar su homeostasis incluyen la información genética estructural de sus células somáticas (heredadas de sus células germinales) y los elementos genómicos, epigenómicos y proteómicos funcionales adquiridos para vigilar y preservar el estado integral de salud: respuesta inmunológica, metabólica, de reparación de tejidos, de detoxificación, de influencias insalubres medioambientales (hábitos, costumbres y exposiciones inadecuadas etc.). Ambos fenotipos particulares, el de las células tumorales y el de la respuesta sistémica de defensa del organismo, influyen en la capacidad/incapacidad para obtener la compensación biológica sistémica (homeostasis), en la respuesta al empleo de diferentes tratamientos anticancerosos (por ejemplo, medicamentos de quimioterapia, o molecularmente dirigidos), e incluso en el grado de descompensación o de toxicidad secundaria a su empleo terapéutico.
El análisis genómico estructural/funcional de las enfermedades neoplásicas malignas comparado al de otras enfermedades crónico-degenerativas, ha demostrado una gran diversidad de alteraciones en las diferentes poblaciones celulares que conforman un tumor. Actualmente, la secuenciación del genoma total de una célula cancerosa es un proceso factible. Las principales alteraciones genómicas identificadas en las células cancerosas son mutaciones, inserciones, deleciones, cambios en el número de copias de regiones particulares (amplificaciones, deleciones, pérdida de heterocigosidad), re-arreglos génicos, re-arreglos intra e inter-cromosómicos, y alteraciones en regiones no codificantes5. Recientemente, diferentes alteraciones epigenómicas han sido encontradas en las células transformadas, correspondientes a alteraciones en los patrones de metilación de los dinucleótidos CpG, en los patrones postraduccionales y composición de las histonas, en los patrones de RNAs no-codificantes, en el enrollamiento de la cromatina y posicionamiento de los nucleosomas y en los patrones de diferentes complejos proteínicos que regulan la expresión génica6,7.
El estudio del genoma de células cancerosas permite identificar las secuencias nucleotídicas de DNA y/o RNA. Frecuentemente en su estudio, además de analizar la secuenciación nucleotídica de las poblaciones del tumor primario, implica el análisis correspondiente del tejido normal cercano (o del germline-DNA), del tejido estromal (microambiente tumoral) y de poblaciones celulares de sitios de tejidos metastásicos.
Los 3 principales proyectos internacionales enfocados a la caracterización genómica global de las diversas células tumorales son el Cancer Genome Project (CGP), el Cancer Genome Atlas (TCGA) y el International Cancer Genome Consortium (ICGC)8. El CGP se enfocó inicialmente al estudio de las alteraciones somáticas en algunos tumores y líneas celulares tumorales. El TCGA se dirigió al estudio del genoma de 13 tumores, y a partir de 2011 a 25 tipos más. El ICGC es el Consorcio más reciente y tiene como metas, obtener la descripción genómica, transcriptómica y epigenómica de 50 tipos y subtipos de cánceres8. Las secuencias de los genomas de las células cancerosas son verificadas mediante secuenciación repetitiva y luego comparadas con los de células normales similares del mismo tejido, ya que eventualmente las células no-cancerosas también acumulan mutaciones somáticas. La principal meta de la secuenciación del genoma de las células cancerosas es identificar los "cambios o mutaciones conductoras" de la iniciación o progresión tumoral. Estos cambios genéticos conductores de la transformación celular son frecuentemente provocados por la activación de oncogenes o la represión/haploinsuficiencia de TSG9. Además de estos cambios o mutaciones conductoras (aproximadamente entre 10-15 en promedio en cada tipo tumoral), el genoma de las células tumorales presenta "cambios o mutaciones pasajeras" (más de 50), secundarios al efecto de los primeros, pero no relacionados a la carcinogénesis. Así, la exploración integral del genoma de las células cancerosas incluye además de los estudios de genómica estructural, estudios epigenómicos y transcriptómicos5,10.
Los principales cambios o mutaciones conductoras en el cáncer han sido localizados en diferentes familias de genes involucradas en la fosforilación de proteínas y lípidos, los cuales codifican principalmente cinasas, fosforilasas, proteasas, caspasas, etc. Más de 120 genes de cinasas mutadas contribuyen en la oncogénesis, por ejemplo la fosfatidilinositol 3 cinasa (PI3K) que participa en los procesos de proliferación, adhesión, sobrevivencia y motilidad celular11.
La secuenciación de todo el genoma tumoral ha permitido identificar los principales tipos de mutaciones somáticas que se presentan en los tumores malignos, los cuales incluyen cambios en nucleótidos únicos, pequeñas indels, variaciones en el número de copias de regiones intermedias y reorganizaciones cromosómicas por traslocaciones. De ellas, las sustituciones de nucleótidos son las mutaciones somáticas más frecuentes detectadas en los diferentes cánceres y presentan gran variabilidad mutacional entre ellos. En promedio se ha calculado que en cada tumor maligno se presenta una mutación nucleotídica por cada millón de nucleótidos; aunque esta relación difiere ampliamente de varios tipos de tumores; las pequeñas inserciones/deleciones corresponden al segundo lugar, en frecuencia.
En la última década ha mejorado la identificación de las CNVs, y de las reorganizaciones cromosómicas, a partir del análisis del genoma con secuenciadores de segunda generación, principalmente al determinar la secuencia nucleotídica con mejor resolución. Esta mejor tecnología, también ha permitido el estudio del transcriptoma e iniciar el estudio de las vías de señalamientos oncogénicos a partir de emplear como modelo, el análisis mutacional de los diferentes tumores. Particularmente el estudio de las vías de señalamientos oncogénicos ha revelado el concepto de exclusividad mutacional de sólo un gen/proteína en el conjunto de componentes que participan en una vía particular; es decir que por ejemplo en la vía de RAS, sólo KRAS o sólo BRAF sufren mutación, y ambas mutaciones no ocurren usualmente en el mismo tumor. Asimismo aunque las mutaciones "conductoras" (mutaciones no-sinónimas) corresponden a las patogénicas en la transformación, muchas mutaciones "pasajeras" se presentan simultáneamente1,8,9.
En el genoma de las células tumorales, se pueden identificar cambios estructurales génicos de diferentes magnitudes: amplios o mayores a 10 Mgbp, intermedios con rangos de 1Kbpb a centenas de Kbpb, y cambios menores a una Kbpb. Los cambios estructurales más frecuentes corresponden a variaciones en el número de copias de regiones particulares de tamaños menores e intermedias; las variaciones mayores como fusión de genes por translocaciones cromosómicas, son menos frecuentes12. En la última década, los estudios genómicos incluyen además estudios del transcriptoma, micronoma (expresión de microRNAs) y del corte y empalme (splicing) alternativos del RNAm. El primer genoma tumoral completo fue secuenciado en 2008, el cual correspondió a un paciente con leucemia mielocítica aguda, en el cual se demostraron 8 mutaciones en regiones codificantes y 500-1,000 en regiones no codificantes13. En 2010, el primer genoma secuenciado de un tumor sólido fue el de un paciente con melanoma maligno, portador de 33,345 mutaciones de tipo sustituciones (292 en regiones exónicas), cuyo patrón mutacional fue consistente al producido por la exposición a la luz ultravioleta14. En el mismo año, igualmente fueron estudiados los genomas de 2 tipos diferentes de cáncer, de acuerdo a su carcinogénesis, a través de la secuenciación masiva en paralelo. El genoma de las células de un carcinoma mamario de tipo basal junto con sus correspondientes células metastásicas a cerebro y las de un tumor desarrollado por su xenoinjerto en un ratón inmunodeficiente fueron analizados; en este triple modelo, se demostró que las mutaciones encontradas originalmente en el tumor primario, fueron mantenidas en las células metastásicas y en el xenoinjerto, y que un número escaso de mutaciones se agregaron en las células metastásicas y del xenoinjerto, junto con cambios en el número de copias de las mutaciones originales15. En esa misma fecha, el genoma de una línea celular de un carcinoma pulmonar de células pequeñas demostró contener 22,910 sustituciones (134 en regiones exónicas), con el patrón mutacional asociado a la exposición de los carcinógenos del tabaco16.
En los últimos 3 años las determinaciones de los patrones epigenómicos de las células cancerosas han empezado a sumarse a las determinaciones genéticas y genómicas. La factibilidad de secuenciar el genoma de las células cancerosas individuales en un tumor, ha permitido distinguir la heterogeneidad de poblaciones en un mismo tumor, e incluso ocasionalmente, ha sido posible identificar la evolución genómica de sus clonas en condiciones pre y postratamiento17. Basados en los cambios genómicos de las células tumorales, se pueden establecer estrategias terapéuticas personalizadas a través de ensayos clínicos18.
Las principales conclusiones generales obtenidas de los estudios de secuenciación y de expresión génica en las células tumorales son: 1) cada tumor en promedio presenta de 30 a 100 alteraciones estructurales, y la mayoría de ellas corresponden a mutaciones puntuales (sustituciones, indels, amplificaciones); 2) existe heterogeneidad de patrones genómicos dentro de las poblaciones de un mismo tumor; 3) los tipos de mutaciones, transición (sustitución de una pirimidina por otra, o de una purina por otra) y transversión (sustitución de una pirimidina por un purina o viceversa), difieren entre los diferentes tipos de tumores; 4) algunos tumores sólidos en niños presentan menor cantidad de alteraciones génicas1,11.
Una nueva taxonomía de los tumores se está escribiendo al emplear como criterios sus perfiles genómicos. Esta determinación integral genómica mejorará el diagnóstico clínico y podrá ser usado en las estrategias terapéuticas personalizadas; y aunque la aplicación de estos agentes anticancerosos podrá generar resistencia secundaria al tratamiento, debido a la inestabilidad genómica inherente a las células tumorales, como ha sucedido en el caso del empleo del imatinib, los cambios genotípicos de esta resistencia podrán ser potencialmente identificados, para rediseñar la siguiente estrategia terapéutica molecular18.
Biomarcadores moleculares clásicos y no clásicos en Oncología
El análisis molecular de biomarcadores en Oncología se encuentra en progreso, sin embargo la incorporación de ellos a la práctica clínica requiere aumentar nuestro entendimiento de los cambios genéticos y epigenéticos que conducen a la malignidad, y comprobar su validación de utilidad clínica. Algunos marcadores moleculares son actualmente empleados como marcadores diagnósticos, pronósticos, predictivos (dependiente del tratamiento), y asociados al diagnóstico (implican diferentes tipos de asociación). Algunos ejemplos de marcadores "clásicos" de diagnóstico (MD) son la inmunofenotipificación en linfomas no-Hodgkin, la hibridación in situ para determinar la presencia del cromosoma Philadelphia en leucemia mielocítica crónica; de marcadores pronósticos (MP), la determinación de mutaciones de p53 en diferentes cánceres; de marcadores predictivos (MPe) la determinación del receptor-2 del factor de crecimiento epidérmico (HER2-ERBB2) en cáncer mamario; y marcadores asociados al diagnóstico (MAD), como la mutación V600E de BRAF en melanomas. Algunas agencias gubernamentales como la Food and Drug Administration (FDA), en Estados Unidos de Norteamérica se encargan de la regulación del empleo de pruebas de diagnóstico molecular en la práctica clínica.
A continuación se mencionan algunos biomarcadores moleculares identificados en 6 tipos de neoplasias malignas, que a través de estudios clínicos han demostrado niveles altos de evidencia en la asociación diagnóstica o pronóstica19. En gliomas: codeleción de 1p/19q (MD), mutación de IDH (MP), metilación de MGMT (MP y MPe); en cáncer mamario: expresión del receptor estrogénico ER-a), expresión de 21 y 70 genes en las pruebas Oncotype y MammaPrint (MADs) y células tumorales circulantes (CTC) en sangre como MP en pacientes con enfermedad metastásica20; en cáncer colónico: mutaciones de KRAS (MPe), inestabilidad microsatelital por defectos en la reparación del DNA (MAD), la molécula 5-de adhesión celular relacionado con el antígeno carcinoembrionario (MAD), mutaciones de BRAF y KRAS (MADs), CTC, ColoPrint (microarreglo simplificado de expresión génica de 18 genes asociados al pronóstico de pacientes con cáncer colorrectal en etapas I-III)21 e infiltración de linfocitos en el microambiente tumoral (MADs); en cánceres broncogénicos de células no-pequeñas: mutaciones del dominio tirosina-cinasa del receptor del factor de crecimiento epidérmico y de KRAS (MPes), y fusión del gen ALK (MPe); en cáncer de próstata: el antígeno prostático específico-PSA (MD), el RNA no codificante PCA3 (MD) y CTC (MP)22; y en leucemia mielocítica aguda: mutaciones de NPM1, CEBPA, KIT, FLT3, WT1 (MPes)19.
En diversos tipos de neoplasias malignas han sido detectadas diferentes alteraciones cromosómicas totales (aneuploidía de diversos tipos) o segmentarias (deleciones de tipo homocigotas/hemicigotas, duplicaciones, translocaciones) y cambios en el número de copias de regiones de DNA23, que frecuentemente se asocian a variaciones pronósticas, como en sobrevida postratamiento y riesgo de recaídas. En las células cancerosas han sido identificadas gran cantidad de variaciones somáticas en el número de copias de regiones particulares (CNVs), cuya mayoría afectan a las familias de genes relacionados con la regulación de la apoptosis (por ejemplo, BCL2), de la proliferación y de la vía NF-kB24. Frecuentemente las mutaciones somáticas se localizan en sitios frágiles (regiones que muestran elevadas tasas de ruptura cromosómicas en respuesta a agentes nocivos del DNA), e incluyen amplificaciones, reducciones (hemicigotas/homocigotas) y pérdida de la heterocigosidad. Las amplificaciones generalmente se ubican en genes que contribuyen a la oncogénesis (por ejemplo, MYC, MYCN, ERBB2, EGFR, AKT2, REL, etc.), y las deleciones se localizan en genes recesivos en el desarrollo del cáncer, principalmente TSG (por ejemplo, CDKN2A, PTEN, RB, SMAD4, TP53, NF2, MLH1, etc.)21,24.
El ejemplo más sobresaliente de la utilidad de los ensayos de expresión génica en la práctica clínica oncológica ha sido la determinación de los patrones de expresión de 70 genes, a través de un simplificado microarreglo de RNA, relacionado con el riesgo de desarrollar metástasis distantes en pacientes con cáncer de mama en etapa I (ganglios axilares sin infiltración tumoral), el cual ayuda a decidir la prescripción o no, de tratamiento adyuvante con quimioterapia. Este microarreglo denominado MammaPrint emplea 1,900 sondas de DNA, y demostró su utilidad para predecir la sobrevida de este grupo pacientes. A partir de 2007, esta prueba genómica fue validada para su uso clínico por la FDA25.
Han sido encontradas muchas alteraciones epigenéticas en diferentes cánceres humanos; sin embargo la mayoría de los patrones de las principales alteraciones epigenéticas específicas en cada uno de los 200 tipos de neoplasias malignas, están actualmente siendo exploradas.
En conclusión, conviene remarcar que la secuenciación de los genomas de las células tumorales es sólo el principio de un largo viaje cuya meta es el análisis molecular integral de los procesos de iniciación y progresión del cáncer.
El modelo del cáncer mamario
Uno de los tipos de cáncer estudiados desde el nivel genómico estructural y funcional es el cáncer mamario. El cáncer mamario es una enfermedad heterogénea causada por la acumulación progresiva de alteraciones genéticas, que incluyen mutaciones puntuales, indels, duplicaciones, CNVs y rearreglos y translocaciones cromosómicas; y simultáneamente por la acumulación de cambios transcriptómicos y epigenomómicos.
El cáncer mamario en su mayoría es de presentación esporádica, originado por alteraciones genéticas de las células glandulares mamarias (somáticas) y sólo el 10% de todos los cánceres mamarios se asocian a mutaciones de las células germinales (antecedente familiar). Los genes que heredan la susceptibilidad a desarrollar cáncer mamario se clasifican en 3 tipos de acuerdo a su penetrancia: alta, moderada y baja. Las mutaciones de BRCA1 y BRCA2 son de alta penetrancia, su presentación aumenta el riesgo de 10 a 30 veces de desarrollar cáncer mamario, comparados con la población general; BRCA1 y BRCA2 funcionan como TSG en el proceso de reparación del DNA de recombinación homóloga. otros genes mutados que aumentan en menor grado la susceptibilidad a desarrollar cáncer mamario, son TP53, PTEN, StK11/LKB1 y CDH1; CHEK 2, ATM, BRiP1y PALB2; y FGFR2 y MAP3K1 (grupos que han demostrado, moderada y baja penetrancia)26.
Los cánceres mamarios esporádicos son causados por la acumulación progresiva de 50-80 alteraciones genéticas somáticas27; muchas de ellas resultan de errores en la replicación del DNA, coincidentes con exposición de mutágenos exógenos y endógenos. La secuenciación de los exomas de más de 500 cánceres mamarios han identificado en promedio 30,000 mutaciones somáticas (92% puntuales, 8% indels); de ellas, 70% alteran el marco de lectura, 25% no lo alteran o son silenciosas y 5% se presentan en los genes de los RNAs o alteran el splicing28. Los principales genes mutados en los cánceres mamarios corresponden a PI3KCA,TP53, CDH1, GATA3, CDH1, PTEN y AKT, RB1, MLL3; MAP3K1 y CDKNiB. Las principales vías de señalamientos intracelulares alteradas en los cánceres mamarios incluyen la del interferón, del control del ciclo celular, de las de reparación del DNA, de p53, AKT, TGF-α, Notch, y de factores epidermales de crecimiento y de sus receptores, EGFR, FGF, ERBB2, RAS y PI3K26.
Algunas regiones del genoma se encuentran frecuentemente amplificadas en los cánceres mamarios, entre ellas las regiones 17q12 (que contiene el oncogén HER2), 11q13 (CCDN1), 8q24 (MYC) y 20q13. La mayoría de estas regiones contienen varios genes que son importantes para el metabolismo del DNA y para el mantenimiento de la integridad cromosomal28.
La secuenciación completa del genoma de los cánceres mamarios conduce el diagnóstico más preciso y dirige el tratamiento27. Sus variantes inherentes como CNVs y SNPs, y sus alteraciones somáticas adquiridas (CNAs) se asocian a diferentes patrones de expresión de los genes de transformación en los cánceres mamarios, por efecto cis y trans. Las CNAs actúan en cis, cuando impactan la expresión de las propias células tumorales (en una ventana de 3Mbp), como por ejemplo las deleciones de PTEN, PPP2R2A, MTAP, MAP2K4 y del gen del receptor de estrógenos. Las alteraciones en trans, afectan a genes en otros sitios del genoma, por ejemplo repercuten en diferentes vías de señalamientos, como en la deleción del receptor de linfocitos T, el cual participa en la respuesta inmunológica adaptativa; o en deleción AurKB que participa en el mantenimiento de la estabilidad cromosómica29.
Las diferentes alteraciones en los patrones de expresión de las células de los cánceres mamarios se emplean actualmente como criterios taxonómicos de clasificación, los cuales se asocian a comportamientos biológicos/clínicos diferentes de pronóstico y predicción. A través de estos patrones de expresión, los cánceres mamarios se han clasificado en 4 tipos: tumores luminal A (receptor de estrógeno positivo), luminal B (receptor de progesterona positivo), tumores con amplificación de HER2 y tumores de tipo basal (triple negativo, con expresión de queratinas basales). La mayoría de los estudios han demostrado que los cánceres mamarios de tipo basal cursan con menor tiempo de recaída y peor supervivencia, comparativamente a los de tipo luminal30. La determinación de nuevos parámetros moleculares en los cánceres mamarios han identificado 2 nuevos subtipos, el tipo normal y el tipo con claudinas bajas, así como diferencias en las células tumorales en las vías de presentación antigénica, las cuales se mantienen estrechamente relacionadas con la respuesta inmunológica innata y adaptativa del paciente31,32.
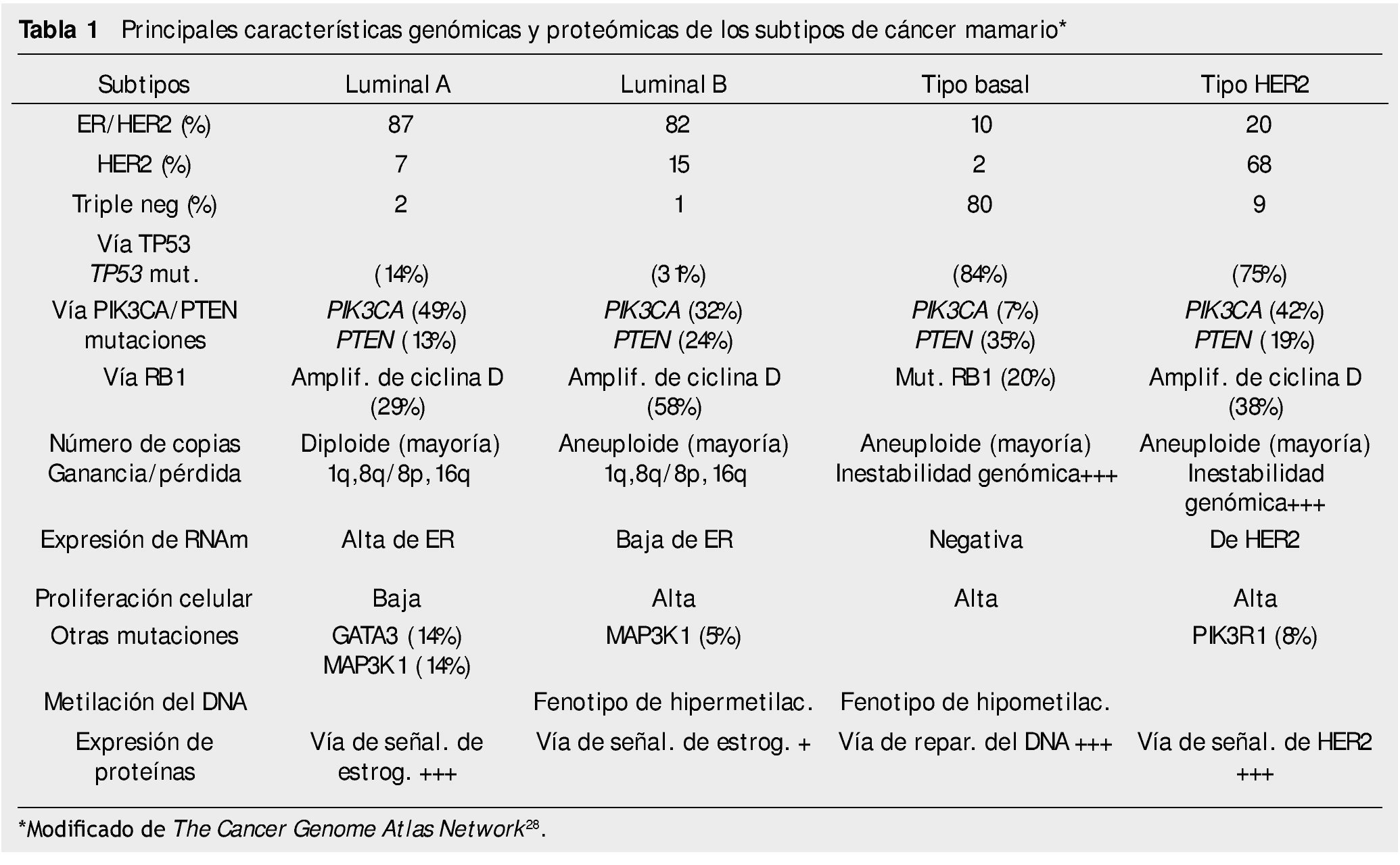
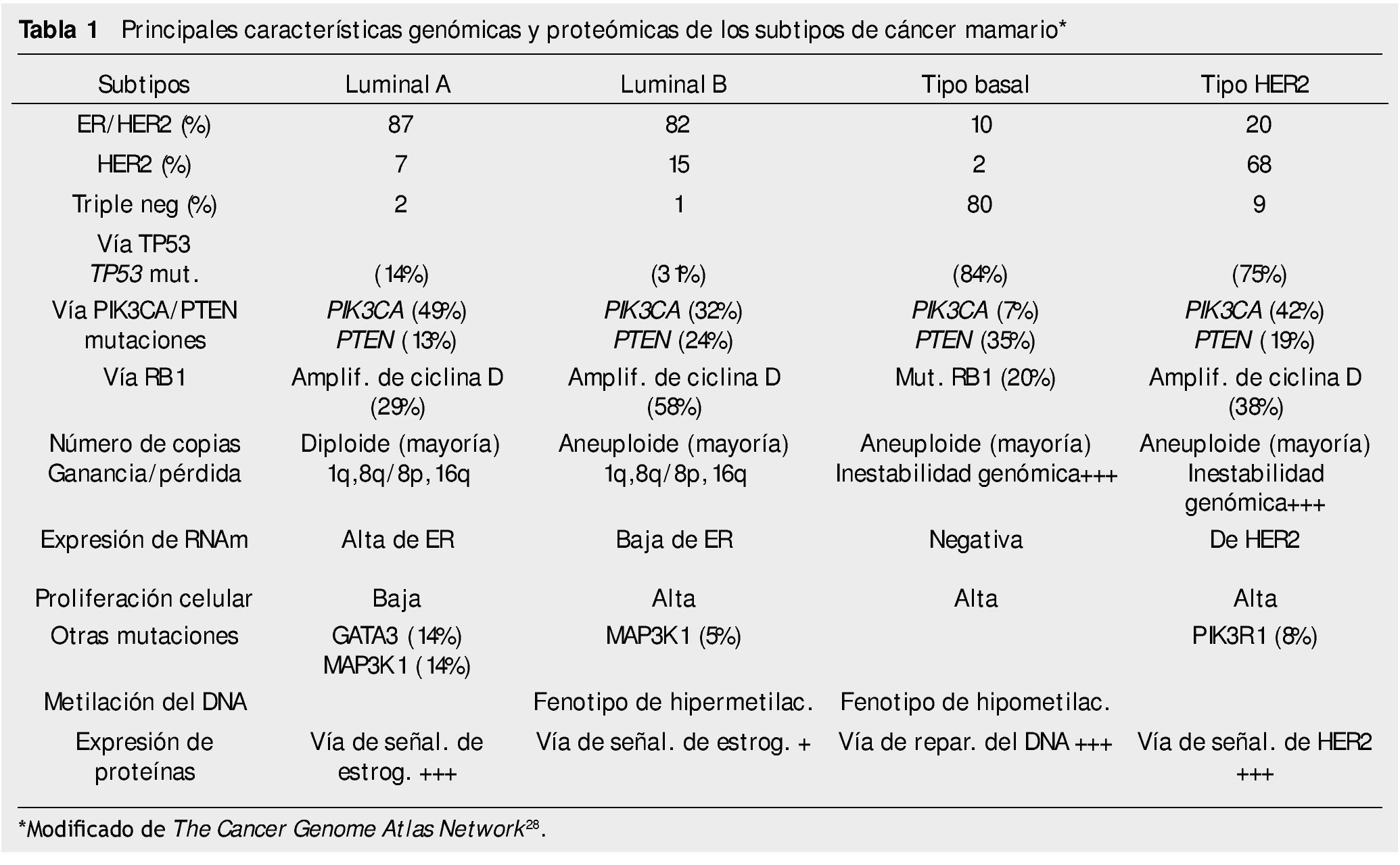
El estudio integral molecular actual de los cánceres mamarios incluye determinar CNVs, CNAs, patrones de metilación del DNA, secuenciación del exoma, patrones de los RNAm, la secuenciación de los microRNAs y la secuenciación y estudio de fase reversa de sus proteínas/fosfoproteínas28. El Cancer Genome Atlas Network ha publicado recientemente un catálogo integral de las principales características genómicas y proteómicas de los 4 subtipos de los cánceres mamarios, que se resumen en la tabla 1.
El entendimiento de los mecanismos reguladores epigenéticos en cánceres mamarios y en general, en la mayoría de los tumores se encuentra en progreso. Desde hace 4 años, se está realizando un gran esfuerzo por múltiples investigadores independientes y aquellos que conforman los grupos del Proyecto Epigenoma Humano y del TCGA33. Actualmente, sólo algunas determinaciones epigenómicas de los cánceres mamarios han sido trasladadas a su aplicación clínica. En particular, el estudio epigenómico de los cánceres mamarios esporádicos, ha identificado frecuentemente el silenciamiento epigenético del gen BRCA1 por la hipermetilación de su promotor34. Recientemente Heyn et al. en un estudio de 15 parejas de gemelas monocigotas con discordancia al desarrollo de cánceres mamarios, identificó 403 diferencias en los patrones de metilación de dinucleótidos CpG de varios de los genes relacionados con la transformación de cánceres mamarios35.
Conclusiones
En las células de los diferentes tipos de cáncer se presentan alteraciones genómicas, epigenómicas y proteómicas. El progreso tecnológico en el estudio genómico ha permitido la determinación de estas alteraciones con mayor resolución picométrica y simultánea y masivamente de sus mediciones en los distintos procesos celulares fenotípicos. Actualmente, muchas investigaciones están dirigidas en la determinación de los perfiles epigenómicos, de las vías de señalamientos oncogénicos, y en la participación de la regulación de expresión génica por las diferentes regiones del DNA nocodificante.
El cáncer mamario es uno de los modelos que ha sido mayormente estudiado desde la perspectiva molecular. Su caracterización en los niveles de genómica estructural, de genómica funcional y proteómico, han permitido construir una nueva taxonomía que los divide en 4 tipos. Esta nueva clasificación complementa a las clasificaciones clínicas y morfológicas, permitiendo aumentar el entendimiento de su biología y concomitantemente el potencial de sus estrategias terapéuticas. El progreso en los estudios moleculares en cánceres mamarios seguirá aumentando nuestro conocimiento de su biología y, sobre su intervencionismo terapéutico y preventivo.
Conflicto de intereses
Los autores declaran no tener ningún conflicto de intereses.
Financiamiento
Los autores no recibieron patrocinio para llevar a cabo este estudio.
* Autor para correspondencia:
Andrés Molina Enríquez N° 361, Colonia Ampliación Sinatel, Delegación Iztapalapa, C.P. 09479, México D.F., México.
Teléfono: (55) 5674 3439.
Correo electrónico: vvaldespinog@yahoo.com.mx (Víctor Manuel Valdespino-Gómez).