



El trasplante de células progenitoras hematopoyéticas (TCPH) es un procedimiento en el cual estas células precursoras son infundidas para restaurar la función de la médula ósea, que puede estar parcial o completamente afectada debido a enfermedades propias de la misma o como consecuencia de una alteración secundaria, incluyendo la aplasia medular ocasionada por altas dosis de quimioterapia o radioterapia administradas para erradicar una neoplasia maligna antes del trasplante.
Durante los últimos 20 años, se han identificado fuentes de células madres alternas a la médula ósea, como las de sangre periférica y de cordón umbilical, las cuales han mostrado tener amplia utilidad clínica y una reconstitución inmunológica adecuada.
Ha mejorado la comprensión del papel del complejo mayor de histocompatibilidad en el trasplante alogénico, y se han desarrollado métodos moleculares de mayor precisión para los donadores y receptores del TCPH. Estos avances junto con el crecimiento del número de donadores no emparentados en los bancos internacionales, así como el incremento de unidades de cordón umbilical, han aumentado la posibilidad de encontrar un donador compatible no emparentado y con ello, la opción de realizar TCPH en un mayor número de pacientes.
En la última década se han ampliado las indicaciones de TCPH y en el futuro sus aplicaciones continuarán aumentando, especialmente en trastornos genéticos. El TCPH se ha convertido en un procedimiento seguro y actualmente se utiliza en forma más temprana en las enfermedades malignas, con lo que es posible obtener mejores resultados cuando este procedimiento está indicado.
Hematopoietic stem cell transplantation (HSCT) is a procedure in which these precursor cells are infused to restore bone marrow function, which may be partially or completely impaired due to a bone marrow disorders or as a result of a secondary failure, including medullary aplasia caused by high doses of chemotherapy or radiation therapy administered to eradicate a malignancy before transplantation.
During the past 20 years, alternative sources of stem cells have been identified, such as are the peripheral and cord blood cells, which have shown broad clinical utility and immune reconstitution adequate.
Our understanding of the major histocompatibility complex role in allogeneic HSCT has improved and more accurate molecular methods for donors and recipients have been developed. These advances, along with the growth in the number of unrelated donors in international banks as well as increasing umbilical cord blood units, have improved the likelihood of finding a donor matched unrelated and thus the option of HSCT in more patients.
In the past decade the indications for HSCT have expanded and their applications will continue to increase in the future, especially in genetic disorders. HSCT is now a safe procedure and is being used earlier for malignant diseases, where it is possible to obtain better results when this procedure is indicated.
Introducción
El trasplante de células progenitoras hematopoyéticas (TCPH) es un procedimiento en el que las células progenitoras hematopoyéticas son infundidas para restaurar la función de la médula ósea (MO), afectada parcial o completamente por enfermedades propias de la MO o como consecuencia de una alteración secundaria.
Antecedentes históricos del TCPH
La historia del TCPH inicia con el concepto propuesto por Arthur Pappenheim en el siglo XIX, de la existencia de una célula precursora de la que se originan todas las células hematopoyéticas. Los trabajos realizados por Lorenz et al. En 1951, mostraron que era posible evitar la muerte de ratones sometidos a dosis letales de radiación, mediante la administración de células de MO de un ratón de la misma cepa1, y en 1956 se demostró que esto era debido a la colonización de la MO del ratón receptor por las células progenitoras hematopoyéticas (CPH) del donador2.
Los primeros TCPH en humanos fueron realizados por E. Donnall Thomas en 19573, quien realizó 6 trasplantes a pacientes con diversas patologías. Las CPH fueron obtenidas de costillas de cadáveres, costillas resecadas de pacientes durante cirugía y mediante la aspiración de crestas ilíacas de pacientes y de donadores sanos. Los resultados fueron pobres, ya que sólo se logró un injerto transitorio en 2 casos, sin embargo esta primera experiencia demostró que es posible administrar cantidades relativamente grandes de MO por vía intravenosa sin toxicidad.
En 1959, Mathé logró llevar a cabo el primer trasplante alogénico4, aunque el paciente falleció por múltiples complicaciones de lo que ahora conocemos como enfermedad injerto contra hospedero (EICH) crónica. En la década de los 60's, Mathé y Thomas intentaron infructuosamente realizar trasplantes alogénicos en pacientes con leucemia aguda usando radiación corporal total (RCT), con dosis de 400-600cGy. Posteriormente, estudios en perros mostraron que se requerían dosis superiores a 800cGy para lograr una inmunosupresión suficiente, que permitiera que la MO alogénica se injertara.
Un descubrimiento crítico en el desarrollo del trasplante alogénico de células progenitoras hematopoyéticas fue el reconocimiento del complejo mayor de histocompatibilidad en humanos (HLA), descrito por Dausset y Payne. Este descubrimiento hizo posible la selección donadores compatibles, que permitieran un injerto duradero sin el riesgo de EICH letal, y permitió además que en 1968, los grupos de Minneapolis y Milwaukee, en forma simultánea, llevaran a cabo los primeros trasplantes exitosos al utilizar MO alogénica de un donador HLA compatible, en niños con inmunodeficiencia grave5,6.
En marzo de 1969, el grupo de Seattle llevó a cabo con éxito, el primer trasplante HLA compatible en un paciente leucémico, empleando RCT y ciclofosfamida (CFM) como esquema de acondicionamiento7. Este mismo grupo publicó en 19728 los primeros 4 casos de anemia aplástica grave (AAG), tratados con trasplante de MO obtenida de donadores HLA idénticos, en los que se empleó CFM como única terapia de acondicionamiento, logrando que 2 de ellos fueran sobrevivientes a largo plazo. Estos estudios demostraron que los pacientes con AAG pueden ser trasplantados exitosamente, y que aquellos con leucemia aguda de mal pronóstico pueden ser curados con TCPH utilizando RCT y CFM.
En México, la historia de los trasplantes de CPH puede dividirse en 2 etapas. La primera inicia en el año de 1980 cuando el Dr. Ricardo Sosa y sus colaboradores llevaron a cabo el primer TCPH en el Instituto Nacional de la Nutrición, en la Ciudad de México. La segunda etapa comenzó a partir de 1995, con el entrenamiento en el extranjero de médicos mexicanos en TCPH.
Una causa que influyó en el desarrollo de los programas de TCPH fue la evolución de los conocimientos en esta área: a) se sustituyó el uso de CPH de médula ósea por CPH de sangre periférica; b) se simplificaron los métodos para llevar a cabo los trasplantes, y c) se iniciaron los alotrasplantes con esquemas de acondicionamiento no mieloablativos9.
Programa de TCPH en el Hospital Infantil de México Federico Gómez
El primer trasplante de CPH del Hospital infantil de México Federico Gómez (HiMFG) fue realizado en octubre de 1989 por médicos del Servicio de Hematología, a una niña de 12 años con diagnóstico de AAG, sin embargo el resultado fue poco favorable y el procedimiento no volvió a realizarse hasta casi 10 años después.
En el periodo comprendido de 1998 a 2005 se realizaron en nuestro Hospital 35 trasplantes de células progenitoras, 11 trasplantes autológos (25.72%) y 24 trasplante alogénicos (74.28%), de los cuales 23 fueron de MO, 4 de sangre periférica y 3 de cordón umbilical, 5 de fuente no especificada; reportándose hasta el momento supervivencia libre de evento de 45% en estos pacientes. Del año 2007 al 2010 se realizaron 9 trasplantes, 6 alogénicos de donador relacionado (66%) y 3 de cordón umbilical (33%), con una supervivencia libre de evento de 56.44.
El programa se interrumpió por más de un año, reiniciando en agosto de 2011, hasta la fecha se han realizado 10 trasplantes más (9 de donador relacionado y uno con doble cordón umbilical), con 2 fallecimientos por complicaciones infecciosas asociadas a EICH grado iv. La supervivencia libre de evento a un año en esta nueva etapa del TCPH en nuestro Hospital, es de 80% a 18 meses.
Objetivos del trasplante de células progenitoras hematopoyéticas
El TCPH tiene 2 objetivos principales:
Sustituir la hematopoyesis del paciente, por ser insuficiente, total o parcialmente defectuosa o neoplásica.
Permitir la administración de un tratamiento antineoplásico intenso con dosis muy elevadas de quimioterapia o radioterapia. En este caso, el TCPH es realmente un recurso de rescate que contrarresta la mielosupresión grave y, es potencialmente mortal del tratamiento antineoplásico.
Para su curación, algunos tumores precisan un tratamiento de intensidad superior a la que se utiliza en los tratamientos estándar, pero inferior a la que origina muerte por toxicidad extrahematológica. Esto originaría mielosupresión prolongada o definitiva, si no se acompañara de la administración de CPH.
Además, en el TCPH procedentes de un donador sano, las células inmunocompetentes derivadas del injerto son capaces de establecer una potente respuesta inmune en contra de las células neoplásicas residuales, fundamento que se conoce como enfermedad injerto contra tumor.
Tipos de trasplante
Dependiendo del origen de las CPH, los trasplantes pueden ser autólogos o alogénicos.
En el trasplante autólogo, los pacientes reciben sus propias CPH, las cuales deben ser cosechadas antes del acondicionamiento. Este tipo de trasplante se realiza preferentemente en tumores sólidos.
En el trasplante alogénico, los pacientes reciben las células madre de un individuo de la misma especie. Puede tratarse de un donador relacionado, que es generalmente un hermano o uno de los padres. Cuando el hermano es un gemelo idéntico, al trasplante se le denomina singénico, y si el donador es el padre o la madre, se le llama haploidéntico. El donador de CPH puede también ser no relacionado, como en el caso del trasplante de cordón umbilical o de individuos registrados en bancos de CPH10.
Fuentes de células progenitoras hematopoyéticas
Médula ósea
las células madre de la MO se obtienen mediante múltiples punciones en ambas crestas ilíacas posteriores. Estas punciones se efectúan bajo anestesia general. Aunque en algunos casos puede realizarse bajo anestesia epidural. La anestesia general es recomendable, ya que permite que el procedimiento sea más cómodo para el paciente y además facilita la labor del médico. La duración habitual de una aspiración de MO es de 2 a 3 horas. El volumen aspirado de MO es entre 10-20 ml por Kg de peso del donador o receptor, dependiendo quien sea más pequeño. niños tan pequeños como de 4 meses, han sido en forma segura y exitosa donadores de MO. La dosis es de 2.5x108 células nucleadas por Kg de peso del receptor. La MO obtenida puede ser administrada inmediatamente al paciente por vía intravenosa o criopreservada para un uso posterior.
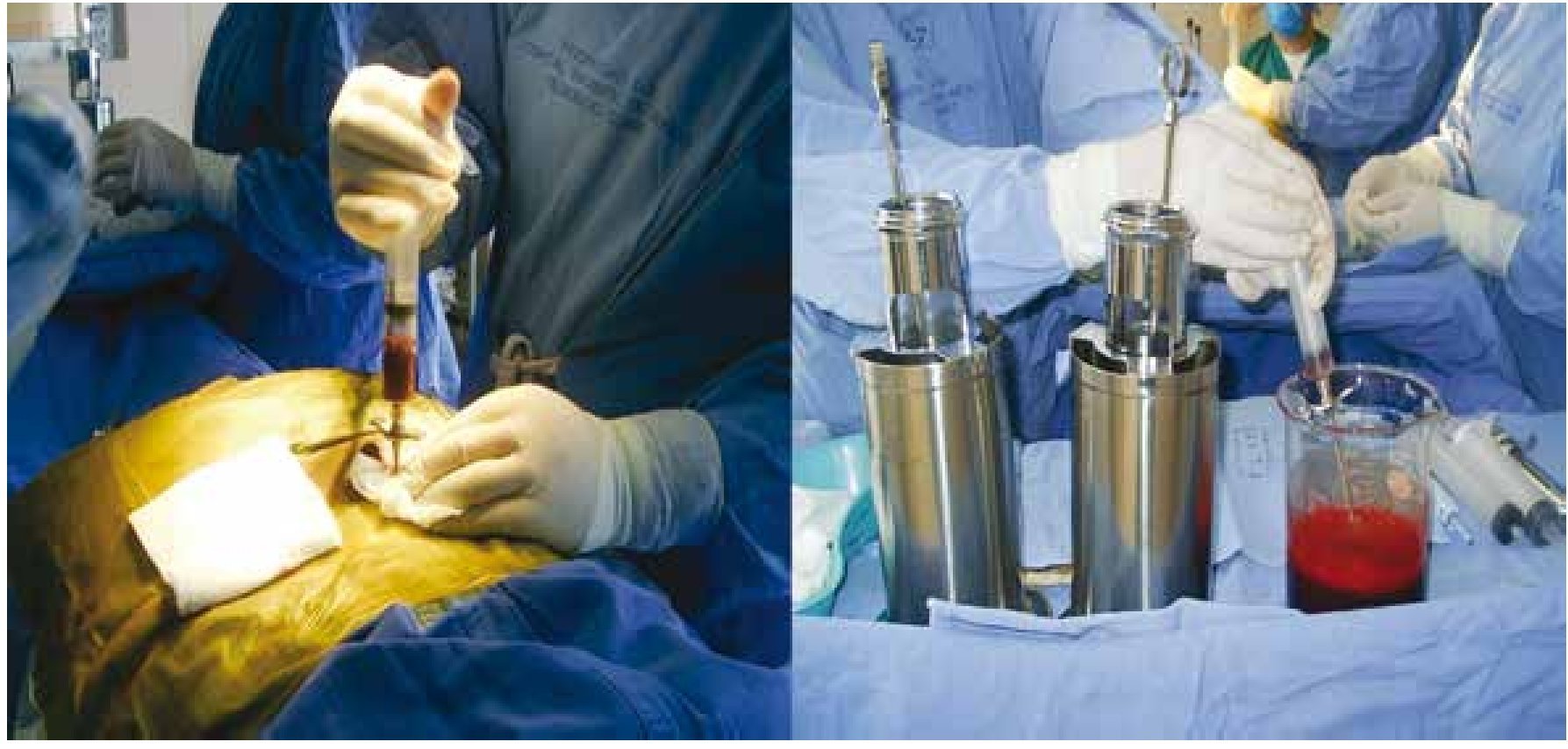
El efecto secundario más frecuente que provoca la donación de MO es el dolor en las zonas de punción, que puede persistir por 24 horas o más y que se controla con analgésicos por vía oral10. En el HiMFG, al igual que en la mayor parte del mundo, el empleo de este tipo de fuente es histórico. La figura 1 muestra la forma en que las CPH eran colectadas de la MO.
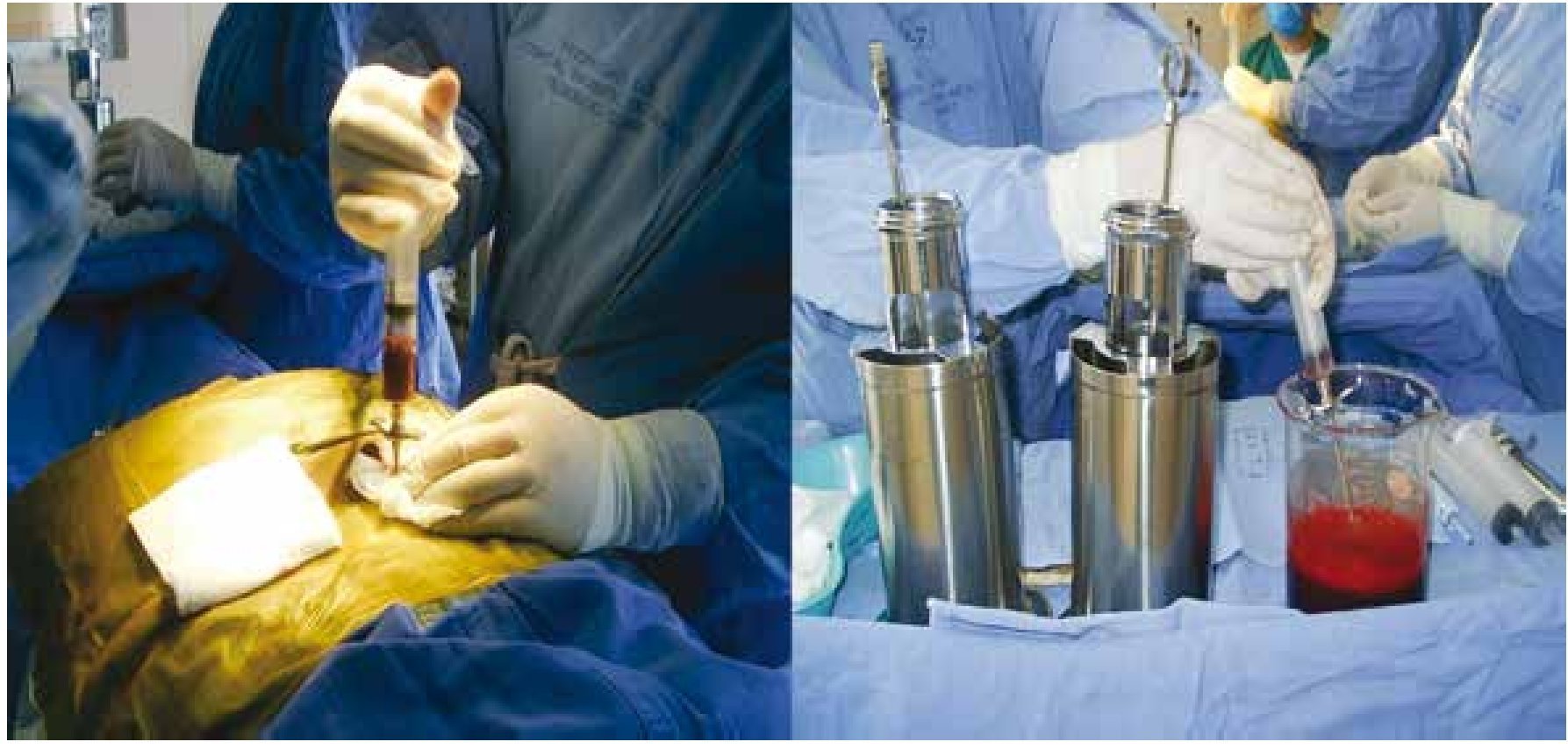
Figura 1. Recolección de células progenitoras hematopoyéticas de médula ósea.
Sangre periférica
Desde 1909 se suponía la presencia de células tallo en la sangre periférica (CTSP), pero la investigación científica para su identificación inició en 1951. Las células progenitoras hematopoyéticas residen en la fracción mononuclear de la sangre periférica, en una concentración de sólo 1% a 10% de las encontradas en MO. Después de la mielosupresión inducida por quimioterapia y del uso de factores de crecimiento, se incrementa el número de progenitores hematopoyéticos en la circulación. La colección de gran número de células mononucleares de sangre periférica es posible gracias al desarrollo de procedimientos de aféresis.
Las CTSP ofrecen ventajas sobre las de MO, lo que ha favorecido el rápido incremento en su uso. Estas ventajas incluyen su recolección rápida y a bajo costo, sin la necesidad de anestesia general; la posibilidad de realizar el procedimiento de manera ambulatoria; su utilización en pacientes con enfermedades que afectan la MO o con antecedente de radiación pélvica; reducción del periodo de citopenias después de la mieloablación, y menor frecuencia de complicaciones infecciosas.
Cordón umbilical
El uso del cordón umbilical (CU) como fuente de células progenitoras hematopoyéticas fue propuesto en 1982, por los doctores Edward A. Boyse, Hal E. Broxmeyer y Judith Bard11,12. El Dr. Broxmeyer experto en hematopoyesis y células madre, junto con el Dr. Boyse, experto e inmunología y genética, consideraron que el CU podría contener células progenitoras, las cuales podían ser utilizadas para TCPH. De esta idea derivaron una serie de estudios realizados in vitro y en ratones para poder demostrar esta teoría13-15. Se realizó un entrenamiento especial entre los ginecoobstetras para una adecuada obtención de la muestras de sangre de CU, las cuales deberían tener un mínimo de contaminación y estar libres de bacterias. Durante este tiempo se crearon un gran número de bancos de cordón para la criopreservación de las células, en nitrógeno líquido.
El primer trasplante de CU fue realizado en Francia en octubre de 1988 por el Dr. Gluckman et al., en un niño de 5 años de edad con anemia de Fanconi16. La sangre de un CU HLA idéntico fue recolectada en Durham, NC, por el Dr. Gordon Douglas, del New York University Medical Center, fue criopreservada en la Universidad de Indiana por el Dr. Brox-Meyers y enviada a París donde el paciente recibió su régimen de acondicionamiento y el trasplante de células de CU. El paciente presentó injerto del donador, y desaparecieron las manifestaciones clínicas de la enfermedad hematológica que padecía.
Los diferentes tipos de células del CU correlacionan con las células presentes en la MO y sangre periférica, sin embargo hay una amplia diferencia en la proporción de estas células y en el contenido de interleucinas. Cairo et al. demostraron una reducción significativa en la producción de la proteína de factor estimulante de colonias de granulocitos, IL3, factor de colonias de macrófagos, factor de crecimiento transformante β1, IL1, IL15 e IL18, además presentaron que hay un incremento en la producción de IL11, factor de células troncales (stem cell factor) y trombopoyetina17.
Indicaciones de trasplante de células progenitoras hematopoyéticas en Pediatría
Enfermedades hematológicas benignas
La AAG es un ejemplo en el cual varios tratamientos están disponibles. El uso de corticoesteroides, andrógenos, globulina antilinfocito (GAL) y ciclosporina A (CsA), solos o en combinación, pueden producir remisión y en ocasiones la curación, sin embargo, aún cuando estos tratamientos sean exitosos existe un riesgo sustancial de mielodisplasia o leucemia aguda subsecuente, especialmente en niños. Además durante estos tratamientos se requiere apoyo transfusional intenso que compromete el éxito de un trasplante posterior. El TCPH alogénico es superior a todas estas alternativas terapéuticas, ya que ofrece con mayor frecuencia una recuperación hematológica persistente17-19.
La anemia de Fanconi (anemia aplásica constitucional) es una enfermedad autosómica recesiva caracterizada por malformaciones congénitas, falla medular y un alto riesgo de desarrollar leucemias agudas y otros tipos de tumores sólidos. La inestabilidad cromosómica es un rasgo característico de estos pacientes y la base para su diagnóstico. Se puede acompañar de malformaciones óseas (ausencia de radio, implantación baja del pulgar, micrognatia), hipopigmentación o hiperpigmentación, estatura baja, microftalmía, malformaciones del tracto urinario (agenesia renal), retraso mental, malformaciones gastrointestinales (agenesia rectal o duodenal), anormalidades cardiacas, sordera, hipogonadismo y síndrome de falla medular20.
Neoplasias hematológicas
Leucemia aguda linfoblástica (LAL)
La LAL puede ser curada con quimioterapia de primera línea en aproximadamente 80% de los casos. El grupo BFM reporta una supervivencia libre de evento de 75.9% a 8 años, en los pacientes incluidos en el estudio BFM90. Resultados similares han sido descritos por diferentes grupos internacionales, como el grupo del Hospital St. Jude. Después de una recaída tardía a MO o extramedular aislada, la supervivencia libre de evento es de 35% a 44%, cuando se emplea quimioterapia convencional21, y este porcentaje es significativamente menor si la recaída ocurre durante tratamiento o en los primeros 6 meses de haberlo terminado de manera electiva.
El trasplante autólogo no ha demostrado mejores resultados que los esquemas intensos de quimioterapia, en pacientes con LLA de alto riesgo en remisión. Este procedimiento podría tener la ventaja de que no produce EICH, sin embargo la respuesta que se puede alcanzar es sumamente limitada, pues aun cuando el paciente se encuentre en remisión medular "completa" antes del trasplante, existe plena evidencia del gran riesgo de injertar células leucémicas en el hospedero y la tasa de recaídas es muy alta.
El papel del trasplante alogénico para paciente pediátricos con LLA ha sido tema de discusión, particularmente en primera remisión, pero si ha mostrado beneficio en pacientes con recaídas tempranas.
El donador relacionado constituye la mejor fuente de CPH, de tal manera que cuando se planea un TCPH, lo primero es investigar a los hermanos del paciente. La fuente de CPH puede ser médula ósea o sangre periférica de donador previamente estimulado. Sin embargo, sólo es posible encontrar un hermano HLA compatible en menos del 20% de los casos que requieren TCPH.
Leucemia aguda mieloblástica (LMA)
El pronóstico de la LMA en niños ha mejorado significativamente en las últimas 2 décadas. Con el uso de quimioterapia intensa, actualmente es posible obtener remisión completa de la enfermedad en 80% a 90% de los casos, y 30% a 70% pueden ser curados si reciben quimioterapia de mantenimiento. El trasplante de donador relacionado en primera remisión permite alcanzar supervivencia prolongada en 45% a 64% de los casos, por lo que representa una muy buena opción para aquellos pacientes con LMA de alto riesgo, cuyas tasas de supervivencia sin TCPH son inferiores a estas cifras.
Leucemia granulocítica crónica (LGC)
De los trastornos mieloproliferativos, la LGC es la variante más común en niños, aunque representa sólo 1% a 3% de todas las leucemias que se presentan a esta edad. La incidencia estimada de Cr Ph+ en los pacientes pediátricos se ha reportado en menos de uno en 100,000 y es menos común en niños menores de 2 años comparados con otros grupos de edad.
Las características clínicas moleculares, citogenéticas, morfológicas de esta enfermedad en los niños, son similares a las del adulto.
Al igual que en adultos, el TCPH alogénico se ha considerado principalmente para aquellos pacientes con LGC Cr Ph+. Debe tenerse en cuenta que aun cuando se tenga un donador relacionado y a pesar de su potencial curativo, hay factores y riesgos para el procedimiento en este tipo de niños, por lo que cada paciente debe ser seleccionado adecuadamente. En un estudio, se evaluaron 314 niños con LG Cr Ph+ entre 1985 y 2011, la supervivencia global y la supervivencia libre de evento a 3 años fueron de 66% y 55%, respectivamente, y en el análisis multivariado los pacientes mostraron tener mejores resultados cuando eran trasplantados en fase crónica vs. crisis blástica, aunque hay que destacar que más de un tercio de los pacientes en fase aguda o crisis blástica estaban vivos y libres de enfermedad a 3 años. La supervivencia libre de evento fue aún mejor en aquellos pacientes pediátricos que se trasplantaron en los primeros 6 meses del diagnóstico, estos resultados difieren de lo reportado en adultos en donde se ha visto que se tienen mejores resultados, si los pacientes se trasplantan en los 12 meses que siguen al diagnóstico22.
Linfomas Hodgkin y no Hodgkin
El pronóstico para niños y adolescentes con Linfomas de Hodgkin (LH) y no Hodgkin (LNH) es en general bueno. No así en aquellos pacientes con enfermedad avanzada, en los que fallan a la primera línea de tratamiento con quimioterapia y radioterapia o en quienes presentan enfermedad recurrente, particularmente si se trata de recaídas tempranas. Estos casos pueden verse beneficiados y tener una supervivencia libre de enfermedad prolongada con un trasplante autológo. Este procedimiento se ha preferido históricamente sobre el trasplante alogénico, debido a la mayor facilidad para realizarlo y a las complicaciones inmunológicas que se presentan en el trasplante alogénico21.
En los últimos años, la introducción de los regímenes de baja intensidad y la disminución de la muerte relacionada al trasplante han permitido revalorar el trasplante alogénico en etapas avanzadas, considerando el efecto injerto contra tumor. En LNH, los factores de riesgo que predicen una mejor supervivencia son el tipo histológicos y el estado de la enfermedad. En el LH, el nivel de DHL pretrasplante representa un marcador de alto riesgo, siendo la supervivencia libre de evento de 42% vs. 0% en pacientes con DHL normal o alta, respectivamente. La tabla 1 muestra las indicaciones del TCPH en linfomas de acuerdo con el Grupo Europeo de Trasplante de Médula Ósea (EBMT).

Tumores sólidos
El trasplante autólogo de MO es un método en el cual se otorgan altas dosis de terapia citorreductiva a pacientes con cáncer. Muchos de los investigadores han demostrado una curva de dosis-respuesta para tumores. La toxicidad hematológica con frecuencia limita la intensidad de muchos de los regímenes quimioterapéuticos disponibles, para el tratamiento en este tipo de enfermedades.
Neuroblastoma
El neuroblastoma (NB) es un tumor de las células de la cresta neural, que dan origen al sistema nervioso simpático. En países anglosajones representa el tumor sólido extracraneal más común de la infancia, con una incidencia anual de aproximadamente 8 casos por millón de niños en los Estados Unidos. Los pacientes con tumor completamente localizado o aquellos con diseminación a tejidos adyacentes o a ganglios linfáticos, tienen un excelente pronóstico cuando se tratan con resección quirúrgica con o sin quimioterapia.
Los pacientes menores de un año de edad al diagnóstico y con estadios I, II o IVS, también tienen un excelente pronóstico. El 40% a 60% de pacientes con estadio III que tienen marcadores biológicos favorables como una sola copia de N-myc e hiperdiploidía, pueden también ser curados con terapia convencional, dependiendo en parte de la posibilidad de resección quirúrgica. Desafortunadamente, entre una tercera parte y la mitad de los neuroblastomas son estadios IV en niños mayores de un año de edad.
A pesar de que las nuevas terapias y regímenes de quimioterapia han incrementado la posibilidad de obtener remisión en pacientes con NB estadio IV, la supervivencia libre de enfermedad a largo plazo se mantiene baja en estos casos.
En los años 50's y principios de los 60's, cuando no se disponía aún de algunos agentes antineoplásicos, la supervivencia libre de enfermedad era de aproximadamente 5%. A finales de los 60's y en los 70's, posterior a la introducción de quimioterapia con múltiples agentes junto con la cirugía y radioterapia, el porcentaje se incrementó 10% a 20%. Asimismo, desde mediados de los años 80's, una terapia multi-modal más agresiva incluyendo cirugía extensa para erradicar el tumor primario y enfermedad macroscópica en otros sitios, además de radioterapia en casos específicos, en combinación con incremento de la dosis de quimioterapia intensa han permitido que los porcentajes de respuesta completa sean de más del 80% en los niños afectados y la supervivencia global se haya incrementado de unos cuantos meses a varios años en muchos pacientes, aunque la supervivencia a largo plazo se mantiene baja.
Dentro de las estrategias investigadas en la última década se encuentra la terapia con muy altas dosis quimioterapia, la cual ha surgido como una esperanza, ya que tanto el porcentaje como la calidad de la respuesta pueden mejorar al incrementar las dosis de algunos de los agentes antineoplásicos, administrados solos o en combinación.
Esta modalidad de terapia intensa con múltiples agentes ha permitido el desarrollo del trasplante autólogo de MO, y más aún el desarrollo de la terapia de soporte, el paratrasplante autólogo, ha permitido escalar las dosis de terapia citotóxica contra NB, lo que ha resultado en una supervivencia libre de enfermedad de 25% a 50% a 2 años, que es superior a la obtenida con quimioterapia convencional.
Sarcoma de Ewing
El sarcoma de Ewing (SE) es un tumor maligno primario de hueso, que se presenta en niños y adolescentes. Los pacientes con enfermedad multifocal o recaída temprana o múltiple, tienen muy mal pronóstico a pesar del uso de cirugía, quimioterapia y radioterapia.
La introducción de terapia multimodal en la última parte de la década de los 60's y principios de los 70's, mejoró el pronóstico. Sin embargo, los pacientes con enfermedad metastásica al diagnóstico, enfermedad localizada, pero extensa o con tumores irresecables del tronco, seguían siendo de alto riesgo de falla al tratamiento y pocos de estos casos podían ser curados. La mayoría respondía inicialmente al tratamiento, pero eventualmente sufría recaída, principalmente en pulmón, hueso o MO y moría con enfermedad diseminada. Los tratamientos mieloablativos con rescate con MO fueron usados por varios investigadores, para mejorar el pobre pronóstico de los pacientes con SE que presentaban estas características.
En 1981, Cornbleet et al. obtuvieron resultados prometedores usando altas dosis de melfalán, con rescate con trasplante de MO. Los pacientes tenían una respuesta inicial buena, pero generalmente recaían si eran tratados cuando la respuesta era sólo parcial. En 1984 el grupo EBMT reportó 35 casos, y de forma similar demostró un porcentaje de respuesta de 66% en los pacientes evaluados. El análisis en 1992 del EBMT europeo, mostró una supervivencia a 2 años de 31% para 14 pacientes con SE metastásico trasplantados en primera remisión completa y una frecuencia de 37% para pacientes trasplantados en segunda remisión completa. Para 28 pacientes trasplantados con enfermedad medible, la supervivencia a 2 años fue de 25%. En recaída, la tasa de supervivencia a 2 años fue de 33% para 19 pacientes con recaída sensible al tratamiento y de 10% para aquellos con resistencia a tratamiento.
Tumores cerebrales
Los tumores del sistema nervioso central (SNC) son un grupo heterogéneo de neoplasias, que representa 16% a 20% de todas las neoplasias en los pacientes pediátricos, siendo únicamente rebasados en frecuencia por la llA en este grupo de edad. La sobrevida a 5 años en pacientes con tumores del SNC, es de aproximadamente 50%. Los tumores más frecuentes son los astrocitomas de bajo grado y los tumores embrionarios como meduloblastoma. Los tumores cerebrales malignos en los niños tienen un mal pronóstico. La sobrevida libre de enfermedad se encuentra entre 40% y 60%, para los tumores más frecuentes (meduloblastoma y ependimomas).
Los pacientes con gliomas de alto grado que progresan después de una terapia inicial tienen un pronóstico muy malo, con una supervivencia media de 7 meses. Gianone y Wolff llevaron a cabo un estudio con 16 pacientes con gliomas del SNC con progresión, previamente tratados con radiación máxima y quimioterapia. Los pacientes entraron a un estudio de fase II con altas dosis de etopósido, seguidas de TCPH autólogo. Tres pacientes (19%) experimentaron respuesta tumoral. Los pacientes respondedores fueron tratados con 2 ciclos más, de altas dosis de etopósido. La media de sobrevida para los 16 pacientes fue de 4 meses, con 3 respondedores que vivieron 9, 10 y 54 meses pos trasplante. Estos resultados correlacionan con los resultados obtenidos cuando se emplean dosis estándar de etopósido. La severa mielosupresión asociada con este régimen y su modesta actividad sugiere que esta alternativa no es eficiente. Deben realizarse más estudios con etopósido solo o en combinación con otros agentes citotóxicos.
Inmunodeficiencias
La inmunodeficiencia severa combinada (SCID) es una alteración en la diferenciación de los linfocitos T. La incidencia estimada es de 1 en 75,000 nacimientos. Algunos investigadores catalogan otras inmunodeficiencias de células T en el grupo de SCID, como por ejemplo la deficiencia de proteína ZAP 70, deficiencia CD3γ, deficiencia en la expresión de HLA clase II, deficiencia de ligasa IV, etc. Esta patología es una emergencia pediátrica, y se necesita un tratamiento inmediato desde que el diagnóstico es confirmado. El tratamiento de elección es el trasplante alogénico, el cual provee la recuperación en aproximadamente 90% de la línea celular deficiente, cuando se lleva a cabo el procedimiento poco después del diagnóstico.
Complicaciones tempranas
Enfermedad injerto contra hospedero
La EICH es la principal causa de morbimortalidad posterior al trasplante alogénico. Es un síndrome clínico derivado de la acción de las células inmunocompetentes del donador contra los tejidos del receptor. Fue inicialmente descrita en modelos murinos como una enfermedad secundaria. Los ratones eran sometidos a radiación y posteriormente transfundidos con células esplénicas normales de otro individuo, desarrollando una serie de manifestaciones a nivel intestinal, hepático y en piel.
La incidencia de EICH grados II a IV es de 40%, pero puede variar desde 10% a 80% según los factores de riesgo. Asimismo, su incidencia varía dependiendo del tipo de trasplante, presentándose en 30% a 50% de los pacientes sometidos a un trasplante de donador relacionado HLA idéntico, y en 50% a 80% de los que reciben trasplante de un donador no relacionado.
En 1996, Billinham formuló 3 requisitos necesarios para la presencia de EICH:
1. Células del donador inmunitariamente competentes.
2. Incapacidad del receptor de destruir o inactivar las células trasplantadas.
3. El receptor debe expresar antígenos que no están presentes en el donador, por lo cual son reconocidos como extraños.
Los principales órganos blancos de la EICH son la piel, hígado e intestinos.
Las manifestaciones más comunes se presentan en piel y se caracterizan por un exantema maculopapular eritematoso, confluente que abarca palmas y plantas. El exantema puede ser asintomático, prurítico o doloroso, y típicamente inicia en zonas expuestas.
La ictericia progresiva es la manifestación hepática más común. En estos casos, la fostasa alcalina es un marcador sensible, pudiéndose encontrar en valores hasta 20 veces mayores que los normales.
A nivel intestinal, se puede manifestar como náusea, anorexia, dolor, sialorrea. En casos graves puede haber daño a la mucosa, con falla de la función intestinal que causa enteropatía perdedora de proteínas con hipoalbuminemia, hemorragia en las evacuaciones o íleo.
Complicaciones tardías
En general, los pacientes con un seguimiento a largo plazo que sobreviven al TCPH conservan una excelente reserva orgánica y pueden incorporarse a las actividades cotidianas propias de la edad. Sin embargo, los esquemas de condicionamiento que contemplan quimioterapia a altas dosis y radioterapia corporal total, conllevan complicaciones que pueden ocurrir inclusive años más tarde.
Entre las complicaciones más graves, sin duda se encuentran las infecciones intercurrentes y la EICH, pero hay otras que deben considerarse en estos pacientes para realizar un diagnóstico temprano, ya que impactan en la morbilidad y mortalidad relacionadas con el TCPH. Las complicaciones se pueden dividir en agudas y crónicas, entre las cuales se encuentran complicaciones hemáticas, cardiovasculares, gastrointestinales, hepáticas, pancreáticas, renales, metabólicas, neurológicas y pulmonares, cuyo tratamiento depende de cada caso23.
El TCPH es un recurso terapéutico de gran utilidad para pacientes pediátricos con diversos padecimientos benignos y malignos no curables por otros métodos.
A pesar de que aún se acompaña de importantes riesgos y de una alta tasa de complicaciones, en Oncología Pediátrica este procedimiento ha permitido mejorar la supervivencia de pacientes de mal pronóstico y en muchos casos representa la única opción curativa, por lo que debe utilizarse siempre que ofrezca un mayor beneficio que el ofrecido por la terapia estándar.
Financiamiento
No se recibió ningún patrocinio para llevar a cabo este artículo.
Conflicto de intereses
Los autores declaran no tener conflicto de intereses.
* Autor para correspondencia:
Dr. Márquez N° 162, Colonia Doctores, Delegación Cuauhtémoc, C.P. 06720, México D.F., México.
Teléfono: 01 55 5228 9917, ext. 2124, 2127, 2130.
Correo electrónico: felixgaytanmorales@gmail.com (Félix Gaytán-Morales).