





La poliposis adenomatosa familiar (PAF) se caracteriza por el desarrollo de cientos a miles de adenomas a nivel del colon y recto. La poliposis adenomatosa familiar atenuada (PAFA) es una forma menos severa, con siempre menos de cien pólipos adenomatosos en el colon y una edad más tardía de aparición1. La PAF tiene un riesgo de malignización del 100%, siendo el único síndrome de cáncer colorrectal (CCR) hereditario en el que queda establecida la necesidad de realizar cirugía profiláctica, y no así, por tanto, en el caso de la PAFA. El debate se establece en cuanto al tipo de cirugía que se debe realizar, según variables del individuo (sexo, edad, deseo reproductivo, preferencias personales), severidad de la enfermedad (gravedad de la afectación rectal, antecedentes de tumores desmoides), o por las complicaciones postoperatorias e impacto en la calidad de vida de los sujetos2,3. La correlación que existe en esta enfermedad entre el genotipo y el fenotipo, también está siendo de utilidad para decidir el tipo de intervención. Presentamos 2 casos que ilustran las distintas opciones quirúrgicas atendiendo a la localización de la mutación en el gen APC.
El primer caso se trata de un paciente de 45 años, con enfermedad por reflujo gastroesofágico, osteocondrosis e hipertensión arterial; sin antecedentes familiares oncológicos de interés. Se le realiza colonoscopia por síntomas abdominales inespecíficos, evidenciándose más de 100 pólipos en todo el marco cólico. En recto se observa un pólipo a 10cm del margen anal, de 1cm de diámetro. Las biopsias resultan de adenomas tubulovellosos con displasia leve, excepto en 3 casos de pólipos localizados en colon izquierdo y transverso, que es severa. El estudio genético identifica mutación germinal de APC (exón 1. c.70C>T). El paciente es intervenido realizándole colectomía total y anastomosis ileorrectal. En la anatomía patológica de la pieza se confirman los múltiples pólipos adenomatosos con displasia leve y moderada en su mayor parte (incluyendo el rectal), excepto en 5, que fue severa.
El segundo caso es una paciente de 28 años, en seguimiento familiar por la unidad de alto riesgo de CCR de nuestro hospital al pertenecer a una familia portadora de mutación en APC (exón 10, c.802G>T), de la cual ella también es portadora. La paciente se encuentra asintomática. Se le realiza colonoscopia, con más de 100 pólipos en todo el colon y el recto, ninguno mayor de 1cm. En esta última localización presenta al menos 12 pólipos. Las biopsias resultan de adenomas con diversos grados de displasia, ninguno de ellos severa. La paciente fue sometida a cirugía profiláctica, realizando proctocolectomía total con reservorio ileoanal. El postoperatorio cursó sin incidencias, confirmándose el diagnóstico endoscópico, sin displasia severa en ninguno de los pólipos.
La PAF es el segundo síndrome hereditario de CCR más frecuente. Se hereda con carácter autosómico dominante, y está causada por mutaciones germinales en el gen APC. Recientemente, también se ha identificado el gen MUTYH, cuya herencia es autosómica recesiva4. La PAF presenta un potencial de malignización muy alto, lo que supone su progresión inevitable a CCR a una edad significativamente más precoz que los casos esporádicos (95% antes de los 50 años). En la PAF pueden asociarse con un amplio espectro de alteraciones en otros órganos. Las manifestaciones extracolónicas más frecuentes son la afectación del tracto digestivo superior. A nivel gástrico, son frecuentes los pólipos de glándulas fúndicas, con un riesgo de desarrollar carcinoma gástrico menor del 1%. Más importancia adquiere la presencia de pólipos duodenales, con una frecuencia del 90% y un alto riesgo de desarrollar carcinoma. Los tumores desmoides son comunes, con un riesgo del 10-30%, siendo la segunda causa más frecuente de mortalidad en la PAF4–6.
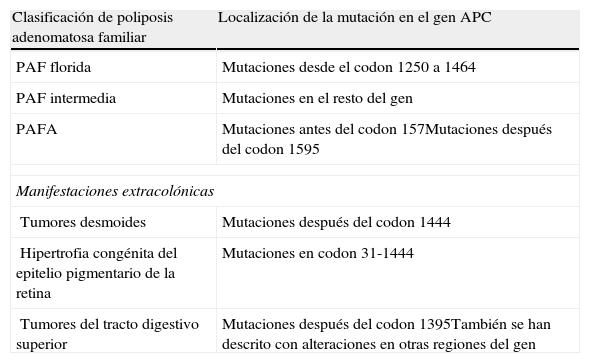
Dada las características del síndrome, se requieren estrategias de manejo diferente, tanto en el seguimiento de los pacientes como en el tratamiento quirúrgico profiláctico2. El debate se establece en el tipo de intervención a realizar, según variables del individuo (edad, sexo, deseo reproductivo, preferencias personales), severidad de la enfermedad y sus manifestaciones extracolónicas, o por las complicaciones quirúrgicas y su impacto en la calidad de vida (infertilidad, incontinencia o urgencia fecal)4. Existen 3 técnicas quirúrgicas principales: proctocolectomía total con ileostomía permanente (PCT), colectomía abdominal total con anastomosis ileorrectal (AIR) y, la más frecuente, la proctocolectomía con reservorio ileoanal (PCR) (tabla 1). Los pacientes que se han sometido a cirugías que no han extirpado por completo la mucosa del colon o con reservorio ileoanal, deben de mantener un seguimiento periódico3,4. Los criterios que hasta el momento se han venido empleando como indicación de proctectomía en estos pacientes, como es la presencia de más de 1.000 pólipos en colon; o más de 20 en el recto, adenomas rectales de más de 3cm, o con displasia severa, pueden ser complementados además mediante el tipo de mutación a nivel de APC que presenten estos pacientes7–9. Numerosos estudios han establecido que parece existir una correlación genotipo-fenotipo de las mutaciones a nivel del gen APC4,9,10 (tabla 2). Así, se ha identificado que los portadores de mutaciones a nivel del codon 1309, se relacionan con un fenotipo más severo, mayor riesgo de pólipos rectales y a una edad más precoz; mientras que mutaciones en el extremo 5’ del gen APC se asocia a formas atenuadas. En los casos que presentamos, por un lado la mutación en APC identifica formas clásicas de poliposis (segundo caso), mientras que en el primero la mutación se puede correlacionar con formas más atenuadas.
Descripción de las principales técnicas quirúrgicas. Ventajas e inconvenientes
| Técnicas quirúrgicas | Ventajas e inconvenientes. |
| Proctocolectomía total con ileostomía permanente | Menos riesgos quirúrgicosProblemas derivados de ileostomíaPeor calidad de vida |
| Colectomía abdominal total con anastomosis ileorrectal | Técnica más sencilla que PCRMenos complicaciones postoperatoriasRiesgo de malignización en el remanenteMejor calidad de vida |
| Proctocolectomía con reservorio ileoanal | Menor riesgo de malignización posteriorTécnica más agresivaDisección pélvica ampliaMayor riesgo de sangrado y lesión de nervios pélvicosMayor riesgo de disfunción sexual e infertilidad en la mujer |
Fuente: Vasen et al.4
PCR: proctocolectomía con reservorio ileoanal.
Resumen de las correlaciones genotipo-fenotipo de la poliposis adenomatosa familiar y sus principales manifestaciones extracolónicas
| Clasificación de poliposis adenomatosa familiar | Localización de la mutación en el gen APC |
| PAF florida | Mutaciones desde el codon 1250 a 1464 |
| PAF intermedia | Mutaciones en el resto del gen |
| PAFA | Mutaciones antes del codon 157Mutaciones después del codon 1595 |
| Manifestaciones extracolónicas | |
| Tumores desmoides | Mutaciones después del codon 1444 |
| Hipertrofia congénita del epitelio pigmentario de la retina | Mutaciones en codon 31-1444 |
| Tumores del tracto digestivo superior | Mutaciones después del codon 1395También se han descrito con alteraciones en otras regiones del gen |
Como conclusión, el mayor conocimiento genético de esta enfermedad favorece un manejo terapéutico más individualizado4,9,10. Los criterios clínicos de indicación del tipo de cirugía pueden ser complementados con criterios moleculares basados en la localización de la mutación en el gen APC.

