




La colangitis esclerosante primaria es una enfermedad colestásica crónica, caracterizada por una inflamación con fibrosis y obliteración de las vías biliares intrahepáticas y extrahepáticas. Se asocia a una colitis ulcerosa en la mayoría de los casos. El proceso de colestasis crónica finalmente conduce a una cirrosis biliar. La enfermedad es poco prevalente en países del sur de Europa y es especialmente frecuente en los países escandinavos. La etiopatogenia es desconocida pero en ella intervienen trastornos de la inmunidad, potenciales agentes tóxicos o infecciosos procedentes del intestino, un daño isquémico de los conductos biliares y quizá una alteración de los transportadores hepatobiliares. Se manifiesta alrededor de los 40 años, predominantemente en varones con clínica y analítica de colestasis. La enfermedad puede ser asintomática. Hay formas específicas de afección de las pequeñas vías biliares intrahepáticas, de predominio en la infancia y síndromes de solapamiento con la hepatitis autoinmunitaria. Se ha descrito una forma caracterizada por un aumento de inmunoglobulina G4 que generalmente se asocia con pancreatitis autoinmunitaria. El procedimiento diagnóstico esencial es la colangiografía retrógrada endoscópica, si bien la colangiorresonancia es el primer procedimiento diagnóstico que debe utilizarse, ya que es igualmente informativa y no invasiva. La biopsia hepática no es esencial para el diagnóstico. La enfermedad es progresiva con una probabilidad de supervivencia libre de trasplante de 18 años en las formas asintomáticas y de 8,5 años en las formas sintomáticas. El colangiocarcinoma es una consecuencia de la enfermedad y comporta un mal pronóstico. No existe un tratamiento específico de la enfermedad, aunque el ácido ursodesoxicólico mejora las alteraciones bioquímicas de colestasis. El trasplante hepático es el último recurso terapéutico con buenas expectativas de supervivencia, aunque con una probabilidad de recidiva de la enfermedad en el hígado trasplantado.
Primary sclerosing cholangitis is a chronic cholestatic disease characterized by inflammation with fibrosis and obliteration of the intra- and extrahepatic bile ducts. This disease is usually associated with ulcerative colitis. The process of chronic cholestasis eventually leads to biliary cirrhosis. The prevalence of primary sclerosing cholangitis is low in southern Europe but is especially high in Scandinavian countries.
The etiopathogenesis is unknown but immune disorders, potential toxic agents or intestinal infections, ischemic injury to the bile ducts, and possibly alterations in hepatobiliary transporters are known to play a role. The disease manifests at the age of approximately 40 years, mainly in men with clinical and laboratory features of cholestasis but may also be asymptomatic.
There are specific forms in which the small intrahepatic bile ducts are involved, mainly affecting children, as well as overlap syndromes with autoimmune hepatitis. A form characterized by an increase in IgG4 has been described, which is usually associated with autoimmune pancreatitis.
The key diagnostic procedure is endoscopic retrograde cholangiography, although magnetic resonance cholangiography is the first diagnostic procedure that should be used since it is equally informative and non-invasive. Liver biopsy is not essential for diagnosis. Primary sclerosing cholangitis is a progressive disease with a probability of transplant-free survival of 18 years in asymptomatic forms and of 8.5 years in symptomatic forms. Cholangiocarcinoma can result from the disease and confers a poor prognosis. There is no specific treatment although ursodeoxycholic acid improves the biochemical alterations of cholestasis.
Liver transplantation is the last therapeutic resort with good results in terms of survival although the disease can recur in the transplanted liver.
La colangitis esclerosante primaria (CEP) es una enfermedad colestásica crónica, caracterizada por inflamación y fibrosis de las vías biliares intrahepáticas y extrahepáticas. Aunque el curso clínico es muy variable, en la mayoría de los pacientes produce una obliteración irregular de los conductos y progresa hasta originar una cirrosis biliar y sus complicaciones. Frecuentemente se asocia a una enfermedad inflamatoria intestinal, especialmente a una colitis ulcerosa1.
El término primario se utiliza para distinguirlo de otras enfermedades que también ocasionan alteraciones colangiográficas similares, como la colangitis bacteriana crónica en pacientes con estenosis de las vías biliares o coledocolitiasis, las lesiones isquémicas de las vías biliares producidas por agentes como formol, alcohol o fluoxuridina, la colangiopatía infecciosa asociada al síndrome de inmunodeficiencia adquirida, la cirugía previa de las vías biliares así como las neoplasias del tracto biliar1–4.
EpidemiologíaExisten pocos datos epidemiológicos. En uno de los más recientes estudios poblacionales se ha establecido una incidencia de 0,9 casos por 100.000 personas/año y una prevalencia de 13,6 casos por 100.000 habitantes, con tasas de incidencia y prevalencia superiores en varones5. Estos datos son parecidos a los reportados en los países escandinavos, donde esta enfermedad es particularmente prevalente y la primera causa de trasplante hepático. Así, en un clásico estudio epidemiológico realizado en Noruega, la incidencia y la prevalencia fueron de 1,3 y 8,5 casos por 100.000 habitantes, respectivamente6. En otros estudios más actuales en Gran Bretaña y Canadá se han establecido incidencias parecidas7–9 (tabla 1). Estos datos contrastan con la menor incidencia de la enfermedad observada en otras áreas geográficas. La CEP es mucho menos prevalente en los países del sur de Europa, como España10 o Italia, o en otras áreas como Asia11. En España se observó un aumento de la incidencia y de la prevalencia entre 1984 y 1988; se pasó de una incidencia de 0,16 a 0,68 casos por millón de habitantes, y de una prevalencia de 0,78 casos a 2,24 casos por millón de habitantes10. Este estudio refería datos de una serie de 43 pacientes diagnosticados en la década de 1980 y, por consiguiente, probablemente no reflejen la realidad actual, ya que la enfermedad es más conocida y existen mejores procedimientos diagnósticos.
Un dato epidemiológico importante es la estrecha asociación de la CEP con una enfermedad inflamatoria intestinal. Así, se ha estimado que entre el 63–90% de los pacientes del norte de Europa y Norteamérica con CEP tienen una enfermedad inflamatoria intestinal, particularmente colitis ulcerosa, pero que esta asociación es menor en España con tasas del 54%, o en países asiáticos con tasas del 20%. Por otra parte, la prevalencia de CEP en pacientes con enfermedad inflamatoria intestinal oscila entre el 2,4–7,5%, lo que permite estimar la prevalencia de la CEP basados en estos datos.
EtiopatogeniaLa causa de la enfermedad es desconocida y se han propuesto diferentes mecanismos.
La potencial patogenia autoinmunitaria se basa en la observación de alteraciones de la inmunidad humoral con presencia en el suero de niveles elevados de inmunoglobulina (Ig) G e IgM, así como la presencia de autoanticuerpos antinucleares, antimúsculo liso, y especialmente los anticuerpos anticitoplasma de los neutrófilos. También se ha descrito un infiltrado inflamatorio por linfocitos T con aumento de la expresión del factor de necrosis tumoral alfa (TNF-α) y cambios en los colangiocitos que expresan citocinas proinflamatorias y profibrogénicas y moléculas de adherencia, así como la expresión aberrante de antígenos de histocompatibilidad de clase ii12.
Otro de los aspectos que apoyaría la base autoinmunitaria es que ciertos antígenos de histocompatibilidad (HLA) se asocian con una mayor susceptibilidad para la enfermedad. Así, se ha descrito una mayor frecuencia de HLA B8, DR3 (HLA-DRB1*0301) y DRw52a en la CEP, al igual que lo que ocurre en otras enfermedades autoinmunitarias13–15. También se ha referido una asociación con el HLA-DR6, que la presencia de HLA-DR4 tendría un efecto protector16, y que la heterocigosidad DR3, DR2 se asociaría a un peor pronóstico de la enfermedad17. Asimismo, se ha indicado que la susceptibilidad genética podría estar determinada por polimorfismos del TNF-α18, y que un cambio de nucleótido en la posición -308 del promotor del TNF-α implicaría una mayor susceptibilidad para la enfermedad, pero relacionado con el haplotipo B8-DR319. Otros estudios han evaluado polimorfismos de MICA, con resultados variables en relación con mayor susceptibilidad o protección para el desarrollo de CEP20,21. Independientemente de la autoinmunidad, también se ha evaluado la relación de polimorfismos de otros genes que confieren susceptibilidad para la CEP o desarrollo de complicaciones. Así, el polimorfismo del promotor de la estromelisina se ha asociado a mayor gravedad de la enfermedad y quizá a fibrosis, así como a una cierta protección del polimorfismo de ICAM-122.
La patogenia autoinmunitaria se cuestiona por la ausencia de algunas características que la sustentarían, tales como: a) mayor prevalencia de CEP en varones, al contrario de lo que ocurre en las enfermedades consideradas de patogenia autoinmunitaria; b) falta de respuesta terapéutica de la enfermedad a fármacos inmunomoduladores, y c) ausencia de autoanticuerpos específicos, pues los p-ANCA, aunque prevalentes, no son específicos de la enfermedad, y se consideran un epifenómeno.
Debido a la estrecha asociación entre CEP y colitis ulcerosa se ha especulado que la inflamación crónica del colon podría favorecer el paso de bacterias intestinales o de moléculas originadas en el colon directamente al tracto portal y, en consecuencia, se produciría una inflamación crónica de las vías biliares que daría lugar a la enfermedad colestásica. En este sentido, se ha demostrado bacteriemia portal en pacientes con colitis ulcerosa, y también una cierta asociación con H. pylori23 y anticuerpos anti-Chlamydia24. Algún estudio experimental sustentaría la hipótesis de la infección procedente del colon25 pero no existen datos suficientemente sólidos. El papel de los ácidos biliares tóxicos procedentes del intestino es otra de las hipótesis que se ha propuesto, quizá generada en mayor magnitud por la enfermedad inflamatoria intestinal.
La lesión isquémica de los conductos biliares es otra de las hipótesis que se ha planteado para entender la patogenia de la CEP, y se sustenta en la observación de lesiones idénticas a la CEP en pacientes con traumatismos hepáticos con disminución del flujo arterial hepático, en pacientes trasplantados y en pacientes con infusión intraarterial de fluoxiuridina o alcohol.
La observación de que los ratones deficientes en el transportador hepatobiliar Mdr2 (que se corresponde con el transportador MDR3 humano) desarrollen espontáneamente una colangitis esclerosante26 ha suscitado la hipótesis de que la enfermedad en los humanos podría deberse a una mutación genética. Sin embargo, no se ha podido comprobar una alteración de este transportador en pacientes con CEP27, por lo que la contribución de estos mecanismos en la patogenia de la enfermedad sigue siendo incierta. Lo mismo se puede decir del papel del gen de la fibrosis quística, CFTR, con resultados discordantes entre los diferentes estudios28–30.
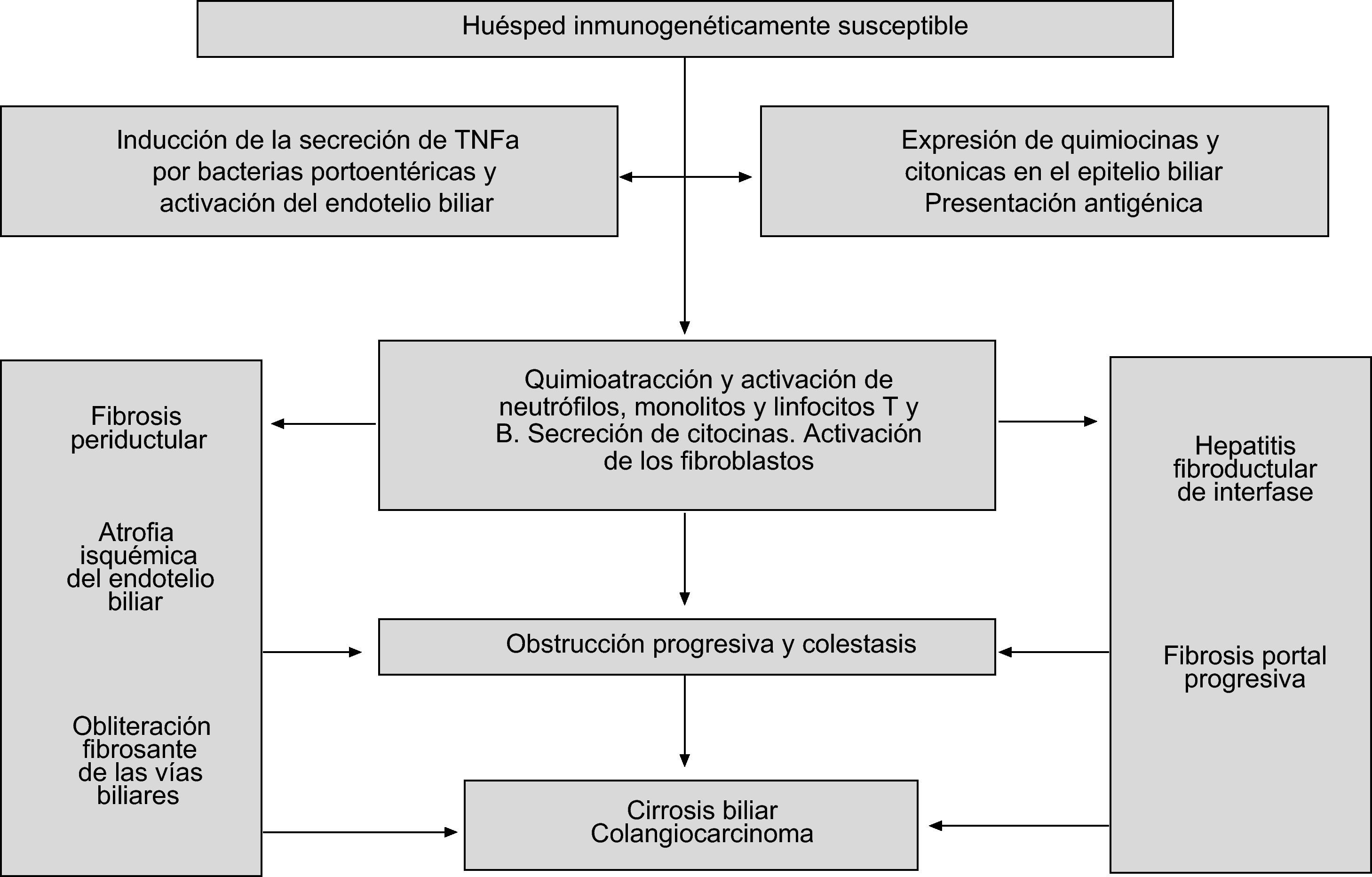
Vierling ha propuesto una hipótesis que engloba casi todos los mecanismos propuestos para entender la patogenia de la CEP (fig. 1). La enfermedad se iniciaría por una respuesta inmunogénica a productos de la pared bacteriana, que daría lugar a la producción de TNF-α31. El aumento de esta citocina a nivel peribiliar atraería neutrófilos, monocitos, macrófagos y linfocitos, e iniciaría el proceso inflamatorio. Consecuentemente, se produciría una fibrosis concéntrica que daría lugar a una atrofia del endotelio biliar secundario a la isquemia. La pérdida paulatina de los conductos biliares produciría colestasis progresiva, fibrosis y cirrosis biliar.
.")
Patogenia de la colangitis esclerosante primaria (adaptado de Vierling et al31).
- 1)
Manifestaciones clínicas y alteraciones analíticas
La enfermedad suele presentarse en varones (70%) de alrededor de 40 años que, además, tienen una colitis ulcerosa. Generalmente transcurren unos 4 años desde la primera alteración bioquímica hasta que se confirma el diagnóstico.
Las manifestaciones clínicas son muy variables32–36. Algunos pacientes carecen de síntomas y se evalúan ante la existencia de un aumento de fosfatasa alcalina, habitualmente en el contexto de una enfermedad inflamatoria intestinal (formas asintomáticas)34. Otros pacientes tienen síntomas inespecíficos e intermitentes más o menos notorios de hepatopatía crónica. En estos casos puede haber signos de colestasis, concretamente prurito, y muy raramente ictericia como primera manifestación. En ocasiones, la enfermedad se diagnostica cuando ya hay una hipertensión portal, con ascitis o hemorragia digestiva por varices esofágicas.
Las determinaciones analíticas revelan un patrón de colestasis con aumento de la fosfatasa alcalina y de la gammaglutamiltransferasa. Una elevación de la fosfatasa alcalina en un paciente con enfermedad inflamatoria intestinal puede indicar el diagnóstico de CEP, pero no es un requisito indispensable, ya que la fosfatasa alcalina es normal en el 8,5% de los pacientes en el momento del diagnóstico de la enfermedad hepática36. También se observa un aumento moderado de las aminotransferasas. En el 60% de los casos la bilirrubinemia es normal en el momento del diagnóstico, así como la albuminemia y la tasa de protrombina. Puede observarse un aumento de las Ig en el 61% de los casos, preferentemente de la IgG. La IgM está aumentada en menos casos (20–45%). También se puede observar un aumento de la IgG4 en el 9% de los casos, aun sin aumento de la IgG total37. Los anticuerpos antimitocondriales son negativos, y, desde el punto de vista inmunológico, el dato más característico es la detección de anticuerpos frente a los neutrófilos (p-ANCA) que se observa en el 26–85% de los casos38. Estos anticuerpos que se hallan en la CEP son distintos de los anticuerpos antineutrófilos que se detectan en los pacientes con granulomatosis de Wegener y otras vasculitis39. En la CEP, así como en la colitis ulcerosa, los ANCA se localizan en la periferia del núcleo de los neutrófilos y, por esto, se ha propuesto que deberían denominarse anticuerpos antinucleares de los neutrófilos. Estos anticuerpos son poco específicos y, en consecuencia, poco útiles para el diagnóstico de la CEP, ya que también se observan en pacientes con colitis ulcerosa y hepatitis autoinmunitaria (HAI).
- 2)
Procedimientos diagnósticos
La colangiografía es el procedimiento imprescindible para hacer el diagnóstico de la enfermedad40,41. Los hallazgos radiológicos son característicos con estenosis difusas y zonas con dilataciones saculares, que adoptan un aspecto arrosariado de los conductos biliares intrahepáticos y extrahepáticos. En la mayoría de los casos hay afección intrahepática y extrahepática. Menos del 25% de los casos únicamente tiene afección intrahepática, y la enfermedad está confinada exclusivamente en las vías biliares extrahepáticas en menos del 5% de pacientes. Se ha descrito afección del cístico y de la vesícula biliar en el 15% de los casos42. También se ha referido afección de los conductos pancreáticos.
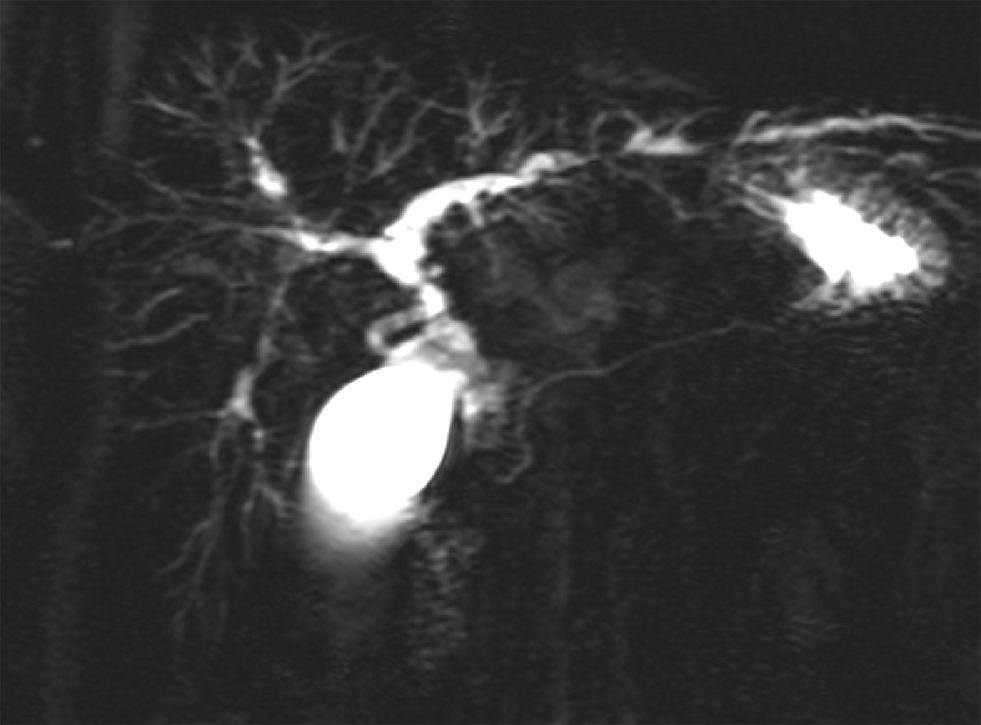
El método colangiográfico más preciso para el diagnóstico es la colangiopancreatografía retrógrada endoscópica (CPRE), pero esta exploración invasiva se asocia a una tasa no despreciable de complicaciones infecciosas y pancreatitis43, por lo que en el momento actual se considera que la colangiorresonancia magnética nuclear (fig. 2) es la técnica inicial y que sólo debe realizarse el procedimiento invasivo (la CPRE) cuando se contempla una actuación terapéutica44. La exactitud diagnóstica de la CPRE y de la colangiorresonancia es similar, pero con esta última se obtiene una mejor imagen de la zona proximal preestenótica a costa de una menor definición de los conductos biliares. La colangiorresonancia alcanza una sensibilidad y una especificidad del 80 y del 87%, respectivamente, para el diagnóstico de CEP41,45. La ecografía tiene escaso interés para el diagnóstico de CEP, salvo para expertos radiólogos que pueden definir dilataciones parciales de los conductos con engrosamiento de la pared.
La biopsia hepática es una prueba adicional para el diagnóstico, que apoya los hallazgos colangiográficos y tiene especial utilidad cuando la enfermedad afecta a los pequeños conductos biliares intrahepáticos. De hecho, las recientes guías clínicas proponen que la biopsia hepática sólo debería indicarse en los pacientes que no tienen evidencias colangiográficas de lesión de grandes vías biliares o cuando hay sospecha de un síndrome de solapamiento CEP/HAI44,46. También es útil para conocer el estadio de la enfermedad. La lesión histológica es una colangitis fibrosa obliterante, definida por una intensa fibrosis concéntrica alrededor de los conductos biliares, que conducen a la obstrucción progresiva y a la sustitución de los conductos biliares por tejido conjuntivo. Esta lesión sólo se observa en el 30% de los casos47. Normalmente, la biopsia hepática muestra cambios inespecíficos de enfermedad biliar, como colestasis, ausencia de conductos biliares en los espacios porta, proliferación periportal de conductos biliares, necrosis parcelar periférica e infiltración inflamatoria de los espacios porta por neutrófilos y linfocitos. Estos hallazgos pueden indicar otros diagnósticos, como la cirrosis biliar primaria, la hepatitis crónica o incluso una obstrucción biliar extrahepática. Las lesiones histológicas de la colangitis esclerosante se han clasificado en 4 estadios47. El estadio i se caracteriza por un aumento del tejido conectivo con agrandamiento de los espacios porta. En el estadio ii, el tejido conectivo aumenta en la zona periportal y hay cambios inflamatorios mínimos. En el estadio iii hay formación de septos fibrosos en el parénquima hepático. Por último, en el estadio iv aparece una cirrosis hepática de origen biliar.
- 3)
Variantes clínicas
- 3.1
Colangitis esclerosante de pequeños conductos
Existe una forma exclusivamente intrahepática de la enfermedad, que representa aproximadamente el 5% de los casos y que se define como colangitis esclerosante de pequeños conductos48–51. En estos pacientes únicamente hay cambios histológicos de la enfermedad, y la colangiografía retrógrada o la colangiorresonancia no muestran las lesiones típicas en las vías biliares intrahepáticas y extrahepáticas. Las manifestaciones clínicas son similares a la enfermedad clásica, y en el transcurso de los años pueden llegar a desarrollar colangitis de grandes vías. Se ha indicado que esta forma representaría una enfermedad con personalidad propia, distinta de la colangitis esclerosante clásica. De hecho, la forma de pequeños conductos tiene un curso más benigno, los pacientes raramente desarrollan un colangiocarcinoma y únicamente una mínima proporción progresa a colangitis de grandes conductos. Asimismo, se ha señalado que esta forma de enfermedad tendría una probabilidad de supervivencia comparable con la de la población general, y mejor que la forma clásica49,52. Este curso, sin embargo, no es constante y algunos pacientes con CEP de pequeños conductos pueden progresar a la forma clásica y requerir, por tanto, un trasplante hepático.
- 3.2
Colangitis esclerosante infantil
En los niños y adolescentes la enfermedad suele presentarse con sintomatología inespecífica, como astenia, anorexia y pérdida de peso53,54. El prurito y la ictericia son menos frecuentes que en los adultos. Los rasgos clínicos más frecuentes, además del aumento de las enzimas de colestasis, son la presencia de una hepatomegalia o una esplenomegalia. La bioquímica tiene cambios mixtos de inflamación y colestasis y, en muchas ocasiones, remeda una HAI, por lo que el diagnóstico sólo puede establecerse mediante colangiografía54.
- 3.3
Colangitis asociada a aumento de inmunoglobulina G4
La colangitis asociada a IgG4 es una enfermedad biliar de etiología desconocida, descrita recientemente, que tiene características bioquímicas y colangiográficas indistinguibles de la CEP. Se caracteriza porque frecuentemente afecta las vías biliares extrahepáticas, responde al tratamiento antiinflamatorio, suele asociarse a pancreatitis autoinmunitaria y otras enfermedades fibrosantes y hay un aumento sérico de IgG4 y un infiltrado de células plasmáticas IgG4-positivas en los conductos biliares y el tejido hepático55–60. No se asocia a enfermedad inflamatoria intestinal. Los datos preliminares indican que la patogenia difiere claramente de las otras enfermedades colestásicas de presunta etiología autoinmunitaria, como la CEP y la cirrosis biliar primaria, ya que en la colangitis con aumento de IgG4 hay una sobreexpresión de linfocitos T cooperadores y T reguladores59. Predomina claramente en varones, y la edad media de diagnóstico es de 60 años58,60.
El tratamiento de elección en esta enfermedad se basa en la administración de corticoides. Se puede asociar azatioprina (hasta 2mg/kg/día) en los casos con estenosis proximal e intrahepática, y tras una recaída durante y después del tratamiento con corticoides. En algunos pacientes pueden ser suficientes 3 meses de tratamiento, pero otros casos requieren tratamiento a largo plazo con dosis bajas de corticoides si no hay remisión completa o hay recidivas.
Para el diagnóstico de esta entidad se han propuesto unos criterios que todavía no están validados y que básicamente consisten en la presencia de una pancreatitis autoinmunitaria, un aumento sérico de IgG4 y una respuesta bioquímica a los corticoides. Sin embargo, en el momento actual todavía hay controversias de si se trata de una nueva enfermedad o bien es una manifestación particular de CEP44.
- 3.4
Síndrome de solapamiento colangitis esclerosante primaria/hepatitis autoinmunitaria
La presencia de un síndrome de solapamiento CEP/HAI se basa en criterios clínicos e histológicos, apoyados por los cambios en la colangiorresonancia o en la CPRE. Esta combinación se ha referido en hasta el 6% de los pacientes con HAI. Cobra especial interés este solapamiento en la infancia, ya que se ha descrito que hasta el 30% de los pacientes pediátricos con CEP tiene rasgos histológicos de HAI, y la enfermedad se manifiesta con una importante hipergammaglobulinemia61,62. Se ha propuesto una denominación específica para esta situación en la que predominan cambios de HAI, conocida como colangitis esclerosante autoinmunitaria63.
La CEP puede aparecer en pacientes que han presentado una HAI previamente, y, en este caso, el síndrome no se correspondería con un solapamiento de ambas enfermedades, sino que serían 2 procesos secuenciales. Este paso de HAI a CEP se describió primero en niños64 y más recientemente se ha publicado una serie de 6 pacientes adultos con HAI que presentaron manifestaciones colangiográficas de CEP al cabo de 5 años del diagnóstico de la primera enfermedad, cuando no habían tenido una buena respuesta al tratamiento inmunosupresor65.
- 3.1

La supervivencia media de los pacientes oscila entre 10–15 años a partir del diagnóstico de la enfermedad32,35,36,66,67. Las formas asintomáticas tienen un mejor pronóstico pero la supervivencia es inferior a la de la población general. La presentación asintomática tiene, no obstante, un curso progresivo con desarrollo de síntomas de colestasis crónica, tales como prurito, molestias abdominales e ictericia, y manifestaciones de hipertensión portal, como esplenomegalia, ascitis y varices esofágicas. En los pacientes sintomáticos, la supervivencia media oscila entre 7–9 años. De hecho, en el estudio pronóstico que ha incluido mayor número de pacientes se ha observado una supervivencia media de 18 años en los pacientes asintomáticos y de 8,5 años en los sintomáticos36.
La edad, los niveles de bilirrubina, la albúmina, las aminotransferasas, la hemorragia varicial, el estadio histológico avanzado y la existencia de enfermedad inflamatoria intestinal se han asociado a un mal pronóstico32,35,36,66,67. Con estas variables se han construido distintos modelos pronósticos. En un estudio multicéntrico realizado en 5 países europeos se describió un modelo pronóstico dependiente del tiempo que permite estimar la probabilidad de supervivencia a corto plazo. Las variables de este modelo son la bilirrubina, la albúmina y la edad en el momento del diagnóstico. La escala de Child-Pugh también parece tener interés para establecer el pronóstico en la colangitis esclerosante68–70. Asimismo, la gradación de las lesiones colangiográficas puede ser válida para establecer el pronóstico de la enfermedad, además de la edad67,71. La existencia de estenosis dominantes igualmente se asocia a un mal pronóstico, y refuerza la importancia de los hallazgos colangiográficos en relación con el pronóstico. La concomitancia con una colitis ulcerosa no condiciona el pronóstico, ya que el curso de la enfermedad hepática es independiente del curso de la enfermedad inflamatoria intestinal acompañante.
En la forma infantil de CEP, los factores relacionados con mal pronóstico son un bajo recuento plaquetar, la esplenomegalia y la mayor edad. Por el contrario, la asociación con un síndrome de solapamiento con HAI o el tipo de tratamiento médico no se han relacionado con menor supervivencia en estas formas, por lo que se ha indicado que aunque el tratamiento farmacológico mejora los síntomas y las pruebas bioquímicas, tiene menor influencia en el curso y pronóstico a largo plazo72.
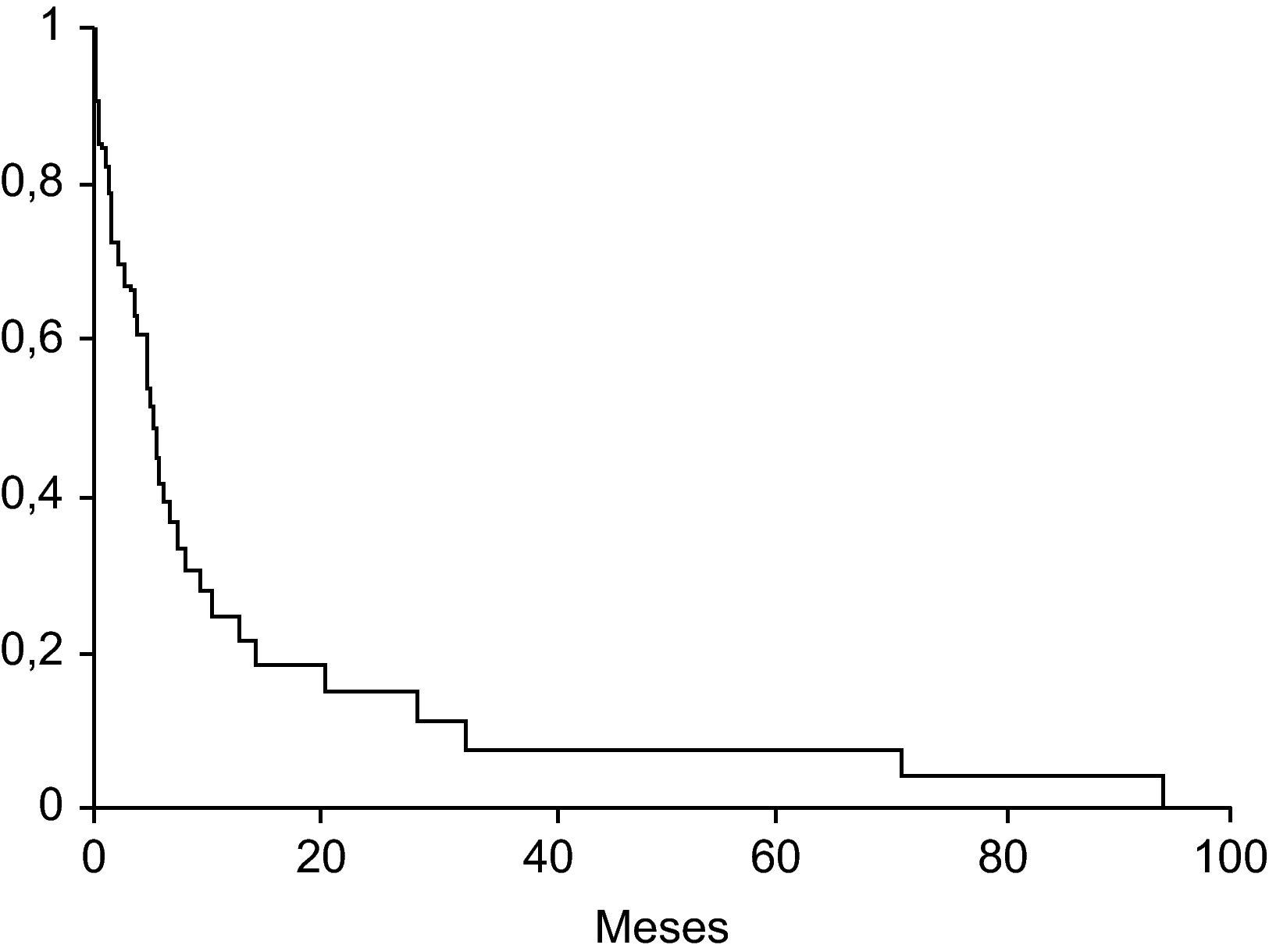
Colangiocarcinoma y otras neoplasiasEntre el 8–18% de los pacientes con colangitis esclerosante desarrollan un colangiocarcinoma, neoplasia que está relacionada con el consumo de tabaco, la coexistencia de enfermedad inflamatoria intestinal, la displasia o el cáncer de colon y la displasia del endotelio biliar73–76. El colangiocarcinoma comporta muy mal pronóstico, con una supervivencia media inferior al año77 (fig. 3). En una elevada proporción de casos la neoplasia se diagnostica durante el primer año de realizado el diagnóstico de la CEP. En una tercera parte de estos casos el tumor puede pasar desapercibido a pesar de utilizar técnicas finas, y se diagnostica en el transcurso del trasplante hepático. La duración de la enfermedad inflamatoria intestinal es un factor asociado a la presencia de colangiocarcinoma77.
.")
Probabilidad de supervivencia en los pacientes con colangitis esclerosante primaria y colangiocarcinoma (Boberg et al77).
Los pacientes con CEP también pueden desarrollar otros tumores, como el hepatocarcinoma, y son más propensos a presentar carcinoma pancreático y carcinoma de la vesícula biliar78,79. En un estudio realizado en Suecia se estimó una incidencia del 1,5% anual para el desarrollo de un carcinoma hepatobiliar79. Asimismo, se ha descrito una mayor susceptibilidad para desarrollar carcinoma colorrectal en los pacientes que tienen una colitis ulcerosa, por lo que estos pacientes requieren un cribado más preciso, con colonoscopias más frecuentes80.
Uno de los problemas del colangiocarcinoma es la dificultad para establecer el diagnóstico, ya que las técnicas de imagen, tanto la ecografía como la tomografía computarizada, tienen una escasa sensibilidad. La técnica más sensible es la colangiorresonancia, aunque sólo detecta el tumor en el 80% de los casos, cuando se asocia a otras técnicas de imagen81,82. Se ha propuesto que la tomografía por emisión de positrones puede ser un buen procedimiento para el diagnóstico de colangiocarcinoma. De hecho, el análisis retrospectivo mostró hallazgos positivos con esta técnica en 3 de 6 pacientes con otras pruebas de imagen negativas81.
Como procedimiento de cribado para el diagnóstico de colangiocarcinoma se ha propuesto la determinación sérica del CEA y del CA 19-9, que tiene una precisión del 86%. El procedimiento es, sin embargo, poco sensible para detectar tumores pequeños83, y en algunos pacientes sin colangiocarcinoma se puede observar un aumento transitorio de estos marcadores. La citología obtenida tras el cepillado de los conductos biliares en el curso de una CPRE es otro procedimiento que aumenta la sensibilidad diagnóstica y se recomienda en los casos con elevada sospecha84–86. El análisis inmunohistoquímico de las mutaciones p53 y K-ras tiene escaso o nulo valor para el diagnóstico de malignidad86. No existen claras pautas de cribado de colangiocarcinoma, pero se ha propuesto que anualmente deberían realizarse una prueba de imagen y una determinación de CA 19-9.
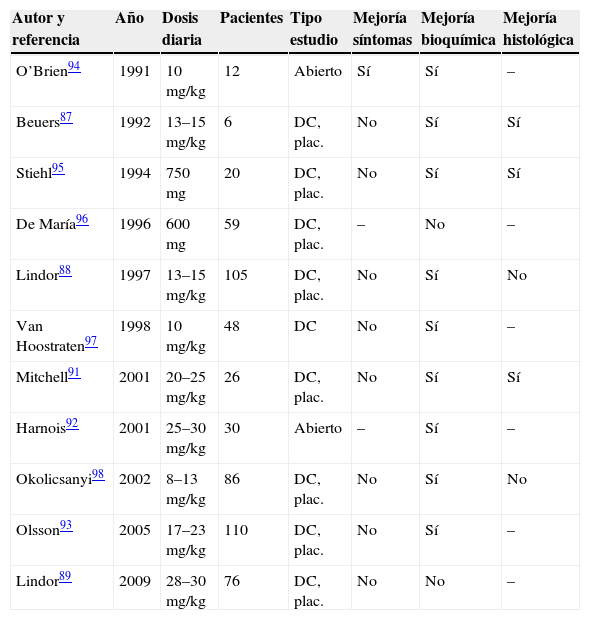
TratamientoEl tratamiento específico consiste en administrar ácido ursodeoxicólico (AUDC) (tabla 2), aunque en la dosis habitual de 13–15mg/kg/día no aumenta la supervivencia ni retrasa la progresión histológica de la enfermedad44,87–99. Existen resultados favorables con dosis de 20mg/kg/día91–93. Co, estas dosis, el tratamiento mejora las alteraciones analíticas pero, sobre todo, retrasa la progresión de la fibrosis y, asimismo, mejora las anomalías colangiográficas. También se ha referido una tendencia a mejorar el proceso inflamatorio91. Estas dosis más altas de AUDC se toleran bien y no se han descrito efectos secundarios, por lo que se han propuesto dosis incluso más elevadas, de 25–30mg/kg/día. Sin embargo, un estudio multicéntrico realizado en EE. UU., en el que se comparaban dosis de 28–30mg/kg/día de AUDC versus placebo, concluyó antes del tiempo previsto debido a una mayor tasa de fallecimiento o trasplante en los pacientes que recibieron el fármaco89. Por consiguiente, estas dosis tan altas son contraproducentes y no deben administrarse a pacientes con enfermedad avanzada.
Tratamiento de la colangitis esclerosante primaria con ácido ursodeoxicólico
| Autor y referencia | Año | Dosis diaria | Pacientes | Tipo estudio | Mejoría síntomas | Mejoría bioquímica | Mejoría histológica |
| O’Brien94 | 1991 | 10mg/kg | 12 | Abierto | Sí | Sí | – |
| Beuers87 | 1992 | 13–15mg/kg | 6 | DC, plac. | No | Sí | Sí |
| Stiehl95 | 1994 | 750mg | 20 | DC, plac. | No | Sí | Sí |
| De María96 | 1996 | 600mg | 59 | DC, plac. | – | No | – |
| Lindor88 | 1997 | 13–15mg/kg | 105 | DC, plac. | No | Sí | No |
| Van Hoostraten97 | 1998 | 10mg/kg | 48 | DC | No | Sí | – |
| Mitchell91 | 2001 | 20–25mg/kg | 26 | DC, plac. | No | Sí | Sí |
| Harnois92 | 2001 | 25–30mg/kg | 30 | Abierto | – | Sí | – |
| Okolicsanyi98 | 2002 | 8–13mg/kg | 86 | DC, plac. | No | Sí | No |
| Olsson93 | 2005 | 17–23mg/kg | 110 | DC, plac. | No | Sí | – |
| Lindor89 | 2009 | 28–30mg/kg | 76 | DC, plac. | No | No | – |
DC: doble ciego; plac.: placebo.
La administración de AUDC en dosis de 15mg/kg/día, además, disminuye de forma significativa el riesgo para desarrollar displasia o carcinoma colorrectal en los pacientes con colitis ulcerosa y CEP100. Se ha publicado que el tratamiento combinado de AUDC con metronidazol (600–800mg/día) durante 3 años se asocia a una disminución significativa del índice pronóstico de la Clínica Mayo e histológica, si se compara con la administración única de AUDC101.
No existen datos consistentes sobre la utilidad de distintos fármacos inmunomoduladores o agentes antifibrosantes pero, en general, los resultados son decepcionantes. Se ha utilizado la azatioprina asociada a prednisona, pero esta última acelera la aparición de osteoporosis y no tiene claros efectos favorables sobre la enfermedad102,103. También se ha utilizado la colchicina sin claros efectos en un estudio doble ciego que incluyó a 85 pacientes104,105. Tampoco se observaron resultados favorables con la d-penicilamina106,107. En otros estudios con menor número de pacientes se ha evaluado el efecto de la ciclosporina y del tacrolimus108,109, pero los datos son poco claros, posiblemente por la duración escasa del tratamiento. Hace años se postuló que el metotrexato podría ser útil110–112, pero en un estudio controlado doble ciego no se observaron efectos favorables en comparación con el placebo. También se ha ensayado la pentoxifilina en un estudio piloto que incluyó a 20 pacientes, en el que se constató la ausencia de efectos favorables tanto en la clínica como en la bioquímica hepática113. Debido a que la nicotina tiene ciertos efectos beneficiosos en la colitis ulcerosa, se ha probado su potencial eficacia en la colangitis esclerosante. Los resultados de un estudio piloto han sido negativos114. También se han evaluado otros agentes solos o combinados con AUDC, con resultados poco claros o ineficaces115,116.
A pesar de los efectos favorables del AUDC, no existen datos claros sobre la supervivencia y, por otra parte, las dosis muy altas pueden ser perjudiciales según el último estudio publicado. No hay datos sobre la potencialidad de otros ácidos biliares, como el 24-norursodeoxicólico, que tiene claros efectos favorables en modelos experimentales117, o el 6-etil-quenodeoxicólico, que recientemente ha mostrado un efecto claramente positivo cuando se combina con AUDC en los pacientes con cirrosis biliar primaria.
Un aspecto que merece especial atención es la actitud ante los pacientes con estenosis biliares únicas o predominantes. Estos pacientes pueden tratarse con dilatación o con prótesis colocada en el interior de la vía biliar118–120. Puede realizarse mediante endoscopia, pero la realización por vía percutánea transhepática puede reducir la incidencia de episodios de colangitis ascendente. En este sentido, existen varios estudios que han demostrado una eficacia de este procedimiento. En un estudio no controlado, la dilatación endoscópica seguida de tratamiento con AUDC parece prolongar la supervivencia cuando se compara con la surpervivencia esperada en un grupo de pacientes no tratados. Otros estudios han mostrado una eficacia similar pero menor tasa de complicaciones en los pacientes con estenosis predominantes tratados con dilatación, en comparación con la prótesis biliar121. A pesar de estos resultados, se aconseja reducir, en lo posible, la manipulación endoscópica o quirúrgica de la vía biliar, ya que estos procedimientos incrementan el riesgo de colangitis.
Asimismo, se aconseja la administración de antibióticos como profilaxis de los episodios de colangitis bacteriana recurrente. Se puede utilizar un solo antibiótico de forma cíclica, como ciprofloxacino, trimetoprima/sulfametoxazol, norfloxacina o ampicilina durante períodos de 3–4 semanas122. Existe una impresión generalizada de que esta actuación es útil, pero no existen estudios controlados que lo corroboren.
La presencia de un colangiocarcinoma, su mal pronóstico a corto plazo y la posibilidad de recidiva en el hígado trasplantado son aspectos que empobrecen la actuación terapéutica en estos pacientes. Recientemente, se están proponiendo medidas muy radicales desde el punto de vista quirúrgico, y también se ha planteado el tratamiento mediante fulguración fotodinámica endoscópica, como un primer paso antes de que se pueda considerar al paciente candidato para el trasplante hepático123. Igualmente, puede contemplarse la radioquimioterapia previa al trasplante hepático con buenos resultados en pacientes muy seleccionados y con el tumor localizado124.
En relación con el trasplante hepático, el problema reside en definir el momento de realizarlo, si bien la mayoría de los grupos están de acuerdo en que las variables que indican el momento del trasplante son los niveles de bilirrubinemia superiores a 6mg/dl durante más de 6 meses, manifestaciones secundarias a la hipertensión portal, como la hemorragia varicial, la ascitis y la encefalopatía hepática, episodios recurrentes de colangitis bacteriana, y prurito refractario al tratamiento médico convencional en ausencia de una estenosis dominante susceptible de corregirse mediante un abordaje radiológico, endoscópico o mixto125. La supervivencia esperable después del trasplante es del 80–90% en el primer año y del 60–80% a los 5 años126,127. En la actualidad, está bien documentada la recurrencia de la enfermedad primaria en el hígado trasplantado con una incidencia que va desde el 5–20%, a partir del primer año del trasplante128,129. La viabilidad del hígado trasplantado y la supervivencia del paciente no están, sin embargo, afectadas negativamente por esta recurrencia. La probabilidad de recurrencia de la CEP después del trasplante está relacionada con la edad del paciente, el sexo masculino, el distinto sexo de receptor e injerto, la coexistencia de enfermedad inflamatoria intestinal, la infección por citomegalovirus y el trasplante con donante vivo129.
El tratamiento sintomático de la CEP es equivalente al de las demás enfermedades colestásicas y consiste en el tratamiento de las complicaciones derivadas de la colestasis, básicamente del prurito, la osteopenia y la malabsorción intestinal. No existen datos recientes que modifiquen los conocimientos actuales sobre el tratamiento de estas complicaciones en la CEP. Sin embargo, se han reportado efectos favorables del alendronato para el tratamiento de la osteopenia de la cirrosis biliar primaria130 y de la diálisis con albúmina como tratamiento extremo del prurito refractario en las colestasis crónicas131.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

