




La enfermedad de Wilson (EW) es un trastorno hereditario que cursa con depósito de cobre (Cu), provocando principalmente clínica hepática, neurológica y/o psiquiátrica. Ante la ausencia de algunos de sus rasgos típicos, el diagnóstico de la EW es difícil y se basa en la combinación de pruebas clínicas, analíticas y genéticas.
El objetivo del estudio fue reflejar la complejidad del diagnóstico de la EW en la práctica clínica.
MétodosSe realizó un análisis retrospectivo de la historia clínica de los pacientes diagnosticados de EW, describiendo la presentación clínica, hallazgos histológicos, analíticos y evolución tras tratamiento. Además se hizo estudio genético y se aplicó el «score» diagnóstico de Leipzig.
ResultadosIncluimos un total de 15 pacientes, 4 sintomáticos: clínica hepática (1), neurológica (1), psiquiátrica (1) y mixta (1) y 11 pacientes presintomáticos: hipertransaminasemia (8) y estudio familiar (3).
Se objetivó anillo Kayser-Fleischer en 2 pacientes, ambos sin clínica neurológica. El 73% presentaba ceruloplasmina ≤5mg/dL y el 40% Cuo 24h>100μg. El Cu hepático superaba los 250μg/g t.s. en el 85% de los pacientes. El estudio genético (mutaciones gen ATP7B) permitió el diagnóstico final en 5 pacientes con mínimos rasgos de la enfermedad, uno de ellos sintomático (clínica psiquiátrica). Se identificaron 5 mutaciones previamente descritas (p.M645R, p.R827W, p.H1069Q, p.P768L y p.G869R) y 3 inéditas (p.L1313R, p.I1311T y p.A1179D), siendo p.M645R la mutación más frecuentemente encontrada.
Tras el tratamiento se objetivó una mejoría de los parámetros analíticos (transaminasas, cupruria) y de la sintomatología, excepto en los pacientes con clínica neuropsiquiátrica.
ConclusionesNuestra serie refleja el papel relevante del estudio genético en el diagnóstico de EW. La identificación en nuestro medio de la mutación p.M645R en la mayoría de nuestros pacientes debe tenerse en cuenta en la estrategia para el análisis molecular del gen ATP7B en nuestra población.
Wilson disease (WD) is an inherited disorder that causes copper (Cu) accumulation, leading to mainly liver, neurological and/or psychiatric manifestations. In the absence of some of the typical features, diagnosis of WD is difficult and is based on the combination of clinical, biochemical and genetic testing. The aim of this study was to illustrate the complexity of the approach to WD in daily clinical practice.
MethodsWe retrospectively analyzed the medical records of patients with WD, including the clinical presentation, histological and biochemical findings, and follow up after treatment. We also carried out genetic testing, and the Leipzig diagnostic score was applied.
ResultsWe included 15 patients. Four were symptomatic, with liver (n=1), neurological (n=1), psychiatric (n=1) and mixed clinical manifestations (n=1), and 11 were presymptomatic, with elevated transaminases (n=8) and family study (n=3).
We observed Kayser-Fleischer ring in 2 patients, both without neurologic symptoms.
Ceruloplasmin ≤5mg/dL was present in 73%, and 24-hour urinary Cu>100μg in 40%. Liver Cu was >250μg/g.d.t. in 85% of the patients. The final diagnosis of WD was given by genetic testing (ATP7B gene mutations) in 5 patients with minimal disease features, including one symptomatic patient (psychiatric symptoms). We identified 5 previously reported mutations (p.M645R, p.R827W, p.H1069Q, p.P768L and p.G869R) and 3 unpublished mutations (p.L1313R, p.I1311T and p.A1179D); the most frequent mutation was p.M645R.
After treatment, biochemical parameters (transaminases, urinary cooper) and symptoms improved, except in patients with neurological and psychiatric manifestations.
ConclusionsOur series illustrates the important role of genetic testing in the diagnosis of WD. The identification of the p.M645R mutation in most of our patients should be kept in mind in the molecular analysis of the ATP7B gene in our region.
La enfermedad de Wilson (EW) es un trastorno hereditario que cursa con depósito de cobre (Cu) en diferentes tejidos, provocando daño hepático, neurológico y/o psiquiátrico. Fue descrita por primera vez en 1912 por Kinnier Wilson. Está clasificada dentro del grupo de las enfermedades raras con una prevalencia variable según las áreas geográficas de aproximadamente 30 pacientes por cada millón de habitantes1,2.
En la práctica clínica diaria y ante la ausencia de algunos de sus rasgos típicos, el diagnóstico de esta entidad es difícil y se basa, generalmente, en una combinación de pruebas clínicas (detección anillo Kayser Fleischer [KF], resonancia cerebral, biopsia hepática), analíticas (ceruloplasmina y Cu sérico y hepático, cupruria) y genéticas (detección de diferentes mutaciones del gen ATP7B), muchas de ellas recogidas en el «score» diagnóstico de EW de Leipzig3,4. Su diagnóstico precoz es importante para la instauración de un tratamiento adecuado, que conlleva en muchos casos, la desaparición de los síntomas y una supervivencia similar a la de la población general.
El tratamiento de la EW se basa en la administración de agentes quelantes de Cu (D-penicilamina, trientina) y/o fármacos que inhiben su absorción intestinal (acetato de cinc). En la actualidad algunos autores desaconsejan el tratamiento con D-penicilamina por su alta toxicidad5.
El objetivo del presente estudio es reflejar la complejidad del diagnóstico de la EW en la práctica clínica habitual, describiendo la presentación clínica y la utilidad del estudio genético y del sistema de puntuación de Leipzig para el diagnóstico de esta enfermedad, en una serie consecutiva de pacientes diagnosticados de EW.
Material y métodosEstudio retrospectivo, descriptivo de una cohorte de pacientes diagnosticados de EW en nuestro medio (hospital terciario). Se revisaron sus historias clínicas recogiendo datos analíticos, clínicos y genéticos.
Como pruebas analíticas, se recogieron hemograma, parámetros hepáticos y de función renal (bilirrubina total, AST, ALT, GGT, FA, albúmina, creatinina), Cu sérico, niveles de ceruloplasmina (en EW habitualmente <20mg/dL), y excreción de Cu en orina de 24h (en EW habitualmente >100μg/24h, en nuestro laboratorio valor normal <80μg/24h). Además se realizó estudio oftalmológico para detectar la presencia de anillo de KF, resonancia magnética (RMN) cerebral para descartar la afectación de ganglios basales y biopsia hepática con cuantificación de Cu en tejido hepático (límite superior de la normalidad [ULN]: 50μg/g tejido seco [t.s.]), en la EW suele ser >250μg/g t.s.
Por último, también se hizo un estudio genético con búsqueda de mutaciones germinales de los exones 2-21 y regiones intrónicas flanqueantes mediante amplificación por PCR y secuenciación automática, y análisis cuantitativo de grandes reordenamientos mediante Multiple Ligation-dependent Probe Amplification (MLPA) con la salsa P098 que contiene sondas para cada uno de los 21 exones y las mutaciones recurrentes (R778L, N1270S, A874V, H1069Q), del gen ATP7B (NM 000053.3).
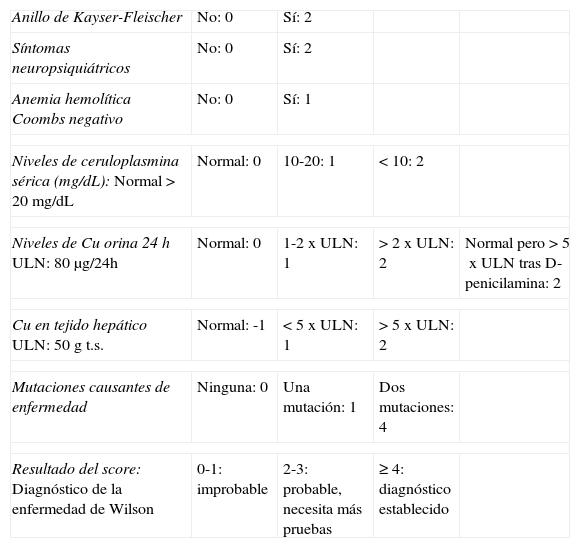
El diagnóstico se realizó con la combinación de pruebas clínicas, analíticas y genéticas. A posteriori, se aplicó el «score» diagnóstico propuesto por «The Working Party at the 8th International Meeting on Wilson’ disease», Leipzig 20013,6 (tabla 1).
«Scoring system» adaptado para el diagnóstico de la EW3
| Anillo de Kayser-Fleischer | No: 0 | Sí: 2 | ||
| Síntomas neuropsiquiátricos | No: 0 | Sí: 2 | ||
| Anemia hemolítica Coombs negativo | No: 0 | Sí: 1 | ||
| Niveles de ceruloplasmina sérica (mg/dL):Normal >20mg/dL | Normal: 0 | 10-20: 1 | <10: 2 | |
| Niveles de Cu orina 24hULN: 80μg/24h | Normal: 0 | 1-2xULN: 1 | >2xULN: 2 | Normal pero >5xULN tras D-penicilamina: 2 |
| Cu en tejido hepáticoULN: 50g t.s. | Normal: -1 | <5xULN: 1 | >5xULN: 2 | |
| Mutaciones causantes de enfermedad | Ninguna: 0 | Una mutación: 1 | Dos mutaciones: 4 | |
| Resultado del score:Diagnóstico de la enfermedad de Wilson | 0-1: improbable | 2-3: probable, necesita más pruebas | ≥4: diagnóstico establecido | |
t.s: tejido seco; ULN: límite superior de la normalidad.
Según la presentación clínica de la enfermedad, los pacientes se clasificaron como sintomáticos o presintomáticos (estudio familiar, hipertransaminasemia asintomática). Los pacientes sintomáticos se diferenciaron a su vez, según la presencia de clínica hepática, neurológica, psiquiátrica o una combinación de ellas.
Los pacientes fueron seguidos en la consulta de hepatología, con control de la sintomatología y monitorización de los parámetros analíticos hepáticos y niveles de Cu en sangre y en orina de 24h. A los pacientes en tratamiento con acetato de cinc se les midieron regularmente los niveles de cinc en sangre y en orina de 24h. Por último, los enfermos con sintomatología neurológica, psiquiátrica y mixta, fueron controlados también por las especialidades de neurología, psiquiatría y medicina interna, respectivamente.
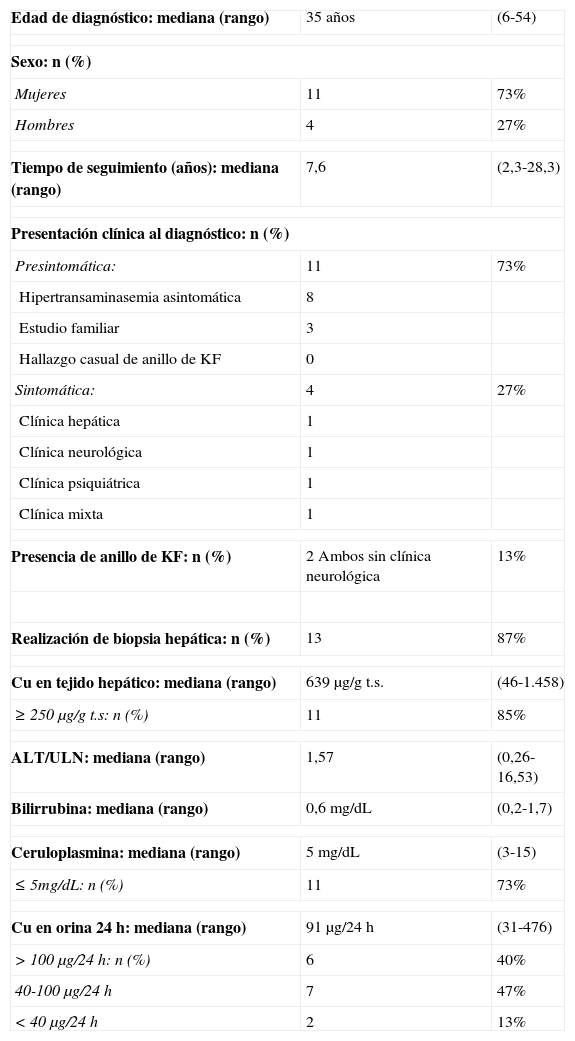
ResultadosSe incluyeron un total de 15 pacientes, 73% mujeres, diagnosticados de EW entre junio 1985 y septiembre 2013 con un seguimiento mediano (rango) de 7,6 años (2,3-28,3). La mediana de edad al diagnóstico fue de 35 años (6-54). La mayoría de los diagnósticos se realizaron en la fase presintomática de la enfermedad, en el contexto de una hipertransaminasemia asintomática. Cuatro pacientes presentaban sintomatología en el momento del diagnóstico: cirrosis y ascitis (P12), parkinsonismo (P13), cuadro psicótico depresivo (P14) y clínica mixta con mialgias-artralgias, astenia, cefalea, cambios de carácter y concentración (P15) (tablas 2 y 3).
Características basales de la población
| Edad de diagnóstico: mediana (rango) | 35 años | (6-54) |
| Sexo: n (%) | ||
| Mujeres | 11 | 73% |
| Hombres | 4 | 27% |
| Tiempo de seguimiento (años): mediana (rango) | 7,6 | (2,3-28,3) |
| Presentación clínica al diagnóstico: n (%) | ||
| Presintomática: | 11 | 73% |
| Hipertransaminasemia asintomática | 8 | |
| Estudio familiar | 3 | |
| Hallazgo casual de anillo de KF | 0 | |
| Sintomática: | 4 | 27% |
| Clínica hepática | 1 | |
| Clínica neurológica | 1 | |
| Clínica psiquiátrica | 1 | |
| Clínica mixta | 1 | |
| Presencia de anillo de KF: n (%) | 2 Ambos sin clínica neurológica | 13% |
| Realización de biopsia hepática: n (%) | 13 | 87% |
| Cu en tejido hepático: mediana (rango) | 639μg/g t.s. | (46-1.458) |
| ≥250μg/g t.s: n (%) | 11 | 85% |
| ALT/ULN: mediana (rango) | 1,57 | (0,26-16,53) |
| Bilirrubina: mediana (rango) | 0,6mg/dL | (0,2-1,7) |
| Ceruloplasmina: mediana (rango) | 5mg/dL | (3-15) |
| ≤5mg/dL: n (%) | 11 | 73% |
| Cu en orina 24h: mediana (rango) | 91μg/24h | (31-476) |
| >100μg/24h: n (%) | 6 | 40% |
| 40-100μg/24h | 7 | 47% |
| <40μg/24h | 2 | 13% |
KF: Kayser-Fleischer; t.s: tejido seco; ULN: límite superior de la normalidad.
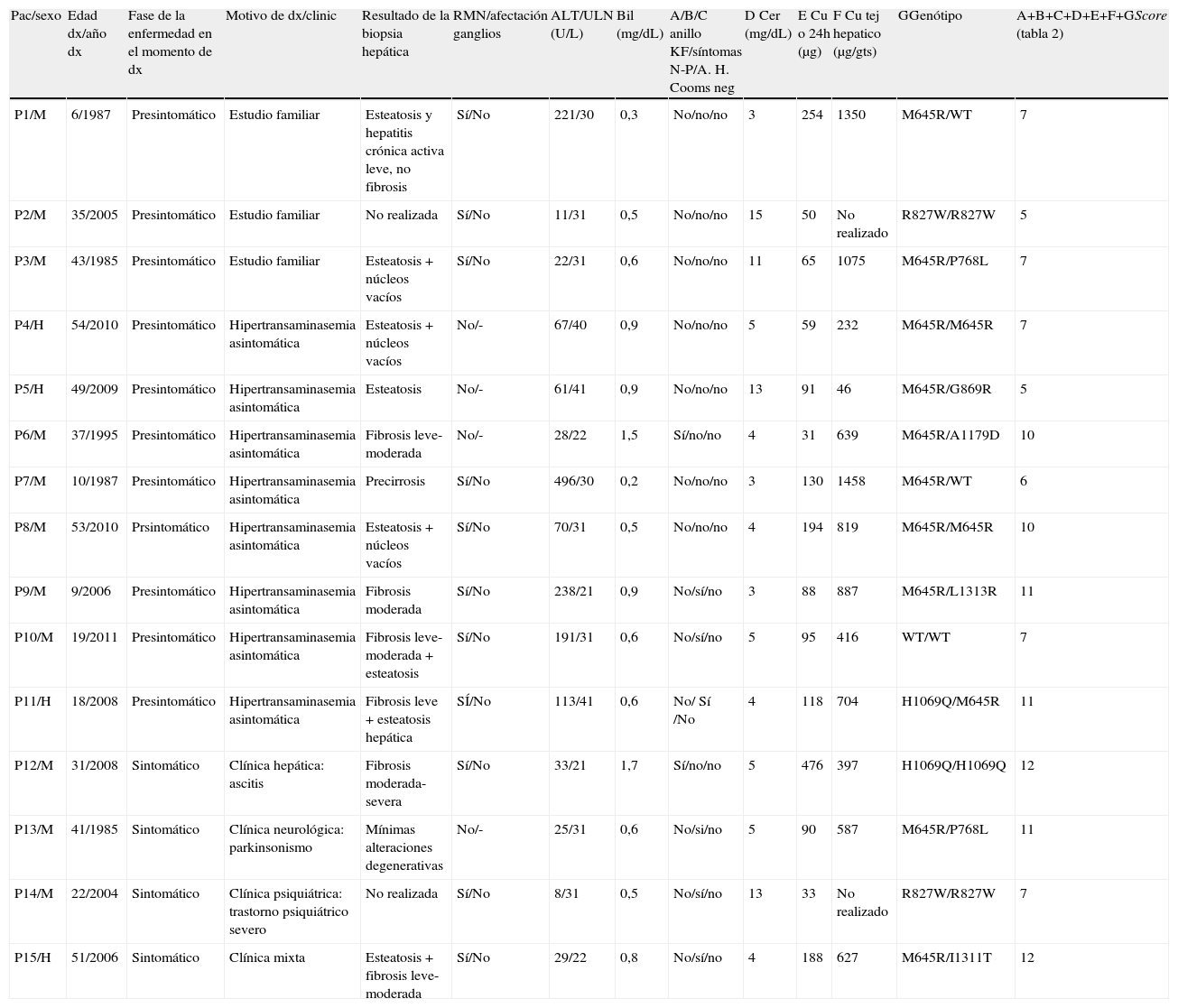
Características clínicas, analíticas y genéticas de los pacientes incluidos en este estudio
| Pac/sexo | Edad dx/año dx | Fase de la enfermedad en el momento de dx | Motivo de dx/clinic | Resultado de la biopsia hepática | RMN/afectación ganglios | ALT/ULN (U/L) | Bil (mg/dL) | A/B/C anillo KF/síntomas N-P/A. H. Cooms neg | D Cer (mg/dL) | E Cu o 24h (μg) | F Cu tej hepatico (μg/gts) | GGenótipo | A+B+C+D+E+F+GScore (tabla 2) |
| P1/M | 6/1987 | Presintomático | Estudio familiar | Esteatosis y hepatitis crónica activa leve, no fibrosis | Sí/No | 221/30 | 0,3 | No/no/no | 3 | 254 | 1350 | M645R/WT | 7 |
| P2/M | 35/2005 | Presintomático | Estudio familiar | No realizada | Sí/No | 11/31 | 0,5 | No/no/no | 15 | 50 | No realizado | R827W/R827W | 5 |
| P3/M | 43/1985 | Presintomático | Estudio familiar | Esteatosis + núcleos vacíos | Sí/No | 22/31 | 0,6 | No/no/no | 11 | 65 | 1075 | M645R/P768L | 7 |
| P4/H | 54/2010 | Presintomático | Hipertransaminasemia asintomática | Esteatosis + núcleos vacíos | No/- | 67/40 | 0,9 | No/no/no | 5 | 59 | 232 | M645R/M645R | 7 |
| P5/H | 49/2009 | Presintomático | Hipertransaminasemia asintomática | Esteatosis | No/- | 61/41 | 0,9 | No/no/no | 13 | 91 | 46 | M645R/G869R | 5 |
| P6/M | 37/1995 | Presintomático | Hipertransaminasemia asintomática | Fibrosis leve-moderada | No/- | 28/22 | 1,5 | Sí/no/no | 4 | 31 | 639 | M645R/A1179D | 10 |
| P7/M | 10/1987 | Presintomático | Hipertransaminasemia asintomática | Precirrosis | Sí/No | 496/30 | 0,2 | No/no/no | 3 | 130 | 1458 | M645R/WT | 6 |
| P8/M | 53/2010 | Prsintomático | Hipertransaminasemia asintomática | Esteatosis + núcleos vacíos | Sí/No | 70/31 | 0,5 | No/no/no | 4 | 194 | 819 | M645R/M645R | 10 |
| P9/M | 9/2006 | Presintomático | Hipertransaminasemia asintomática | Fibrosis moderada | Sí/No | 238/21 | 0,9 | No/sí/no | 3 | 88 | 887 | M645R/L1313R | 11 |
| P10/M | 19/2011 | Presintomático | Hipertransaminasemia asintomática | Fibrosis leve-moderada + esteatosis | Sí/No | 191/31 | 0,6 | No/sí/no | 5 | 95 | 416 | WT/WT | 7 |
| P11/H | 18/2008 | Presintomático | Hipertransaminasemia asintomática | Fibrosis leve + esteatosis hepática | SÍ/No | 113/41 | 0,6 | No/ Sí /No | 4 | 118 | 704 | H1069Q/M645R | 11 |
| P12/M | 31/2008 | Sintomático | Clínica hepática: ascitis | Fibrosis moderada-severa | Sí/No | 33/21 | 1,7 | Sí/no/no | 5 | 476 | 397 | H1069Q/H1069Q | 12 |
| P13/M | 41/1985 | Sintomático | Clínica neurológica: parkinsonismo | Mínimas alteraciones degenerativas | No/- | 25/31 | 0,6 | No/si/no | 5 | 90 | 587 | M645R/P768L | 11 |
| P14/M | 22/2004 | Sintomático | Clínica psiquiátrica: trastorno psiquiátrico severo | No realizada | Sí/No | 8/31 | 0,5 | No/sí/no | 13 | 33 | No realizado | R827W/R827W | 7 |
| P15/H | 51/2006 | Sintomático | Clínica mixta | Esteatosis + fibrosis leve-moderada | Sí/No | 29/22 | 0,8 | No/sí/no | 4 | 188 | 627 | M645R/I1311T | 12 |
A.H: anemia hemolítica; Bil: bilirrubina; dx: diagnóstico; Cer: ceruloplasmina; Cu o 24h: cobre en orina 24h previo a cualquier tratamiento; gts: gramo de tejido seco; H: hombre; KF: Kayser Fleischer M: mujer; N-P: neuropsiquiátricos; ULN: límite superior normalidad; WT: «wildtype», secuencia ATP7B nativa sin mutaciones.
Todos los pacientes presentaban niveles disminuidos de ceruloplasmina, 11 (73%) por debajo o igual a 5mg/dl (tabla 2). La mediana de la excreción urinaria de Cu en 24h fue de 91μg/24h. En 6 pacientes (40%) fue >100μg/24h y en 7 (47%) entre 40-100μg/24h. Dos pacientes (13%) presentaron un excreción urinaria de Cu normal (<40μg/24h) (tablas 2 y 3).
La presencia de anillo de KF se observó en 2 pacientes, uno diagnosticado por hipertransaminasemia asintomática y otro con cirrosis con ascitis. A 11 pacientes (73%) (uno con anillo de KF) se les realizó RMN cerebral, siendo normal en todos ellos.
Se realizó biopsia hepática y cuantificación de Cu hepático a 13 (87%) pacientes. Los hallazgos histológicos más relevantes fueron la presencia de esteatosis y/o fibrosis leve-moderada (tabla 3). El Cu en tejido hepático (mediana-rango) fue de 639 (46-1.458)μg/g t.s., siendo en un 85% superior a 250μg/g t.s (tabla 2).
Se realizó el estudio genético (mutaciones y grandes reordenamientos del gen ATP7B) en los 15 pacientes. Doce (80%) pacientes presentan mutaciones bialélicas, 2 pacientes mutación monoalélica y en un paciente no se identificó alteraciones en la secuencia del gen. Las mutaciones halladas incluyeron 5 cambios previamente descritos (p.M645R, p.R827W, p.H1069Q, p.P768L y p.G869R) y 3 inéditos (p.L1313R, p.I1311T y p.A1179D), con una alta frecuencia de la mutación p.M645R, que fue identificada en el 73% de los pacientes. En la tabla 3 se describen las mutaciones de cada paciente.
Todos los pacientes presentaron un «score» diagnóstico mayor de 4 puntos, límite fijado para establecer el diagnóstico de EW (tablas 1 y 3)3.
La mediana (rango) del «score» de los 15 pacientes fue de 7 (5-12) puntos; 7 (5-11) en pacientes presintomáticos y 11,5 (7-12) en los 4 pacientes sintomáticos en el momento del diagnóstico.
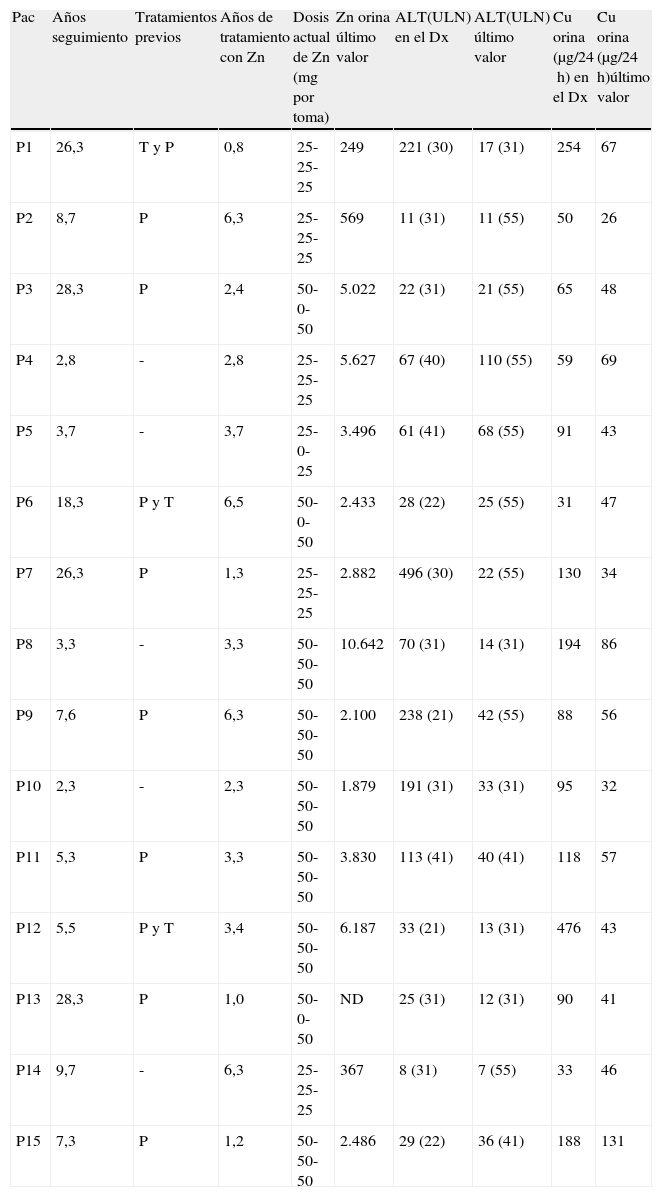
Diez pacientes fueron tratados inicialmente con D-penicilamina y/o trientina (tabla 4). De ellos, 4 pacientes cambiaron el tratamiento a acetato de cinc (cinc) por efectos adversos: intolerancia digestiva (P2), exantema cutáneo (P9), úlceras orales (P11) y hematuria-proteinuria (P12). En los 6 pacientes restantes (2 con suspensión previa voluntaria del tratamiento: P1 y P15) se cambió a tratamiento con cinc por su mejor perfil de seguridad. Al final del seguimiento todos los pacientes seguían tratamiento solo con acetato de cinc. Tres pacientes presentaron en algún momento del seguimiento una excreción urinaria de cinc por debajo 2.000μg/24h, reflejando posiblemente un pobre cumplimiento terapéutico y menos probable una dosis insuficiente de la medicación prescrita (tabla 4). De los 8 pacientes con hipertransaminasemia asintomática, en 6 pacientes (75%) se objetivó una disminución y/o normalización de transaminasas (uno y 5 pacientes respectivamente). De las formas sintomáticas, en una paciente se observó una resolución completa de su sintomatología con normalización de la función hepática tras tratamiento y en los otros 3 pacientes con clínica mixta y/o neurológica –psiquiátrica no se observaron cambios notables en su sintomatología.
Datos de tratamiento y evolución de los valores analíticos
| Pac | Años seguimiento | Tratamientos previos | Años de tratamiento con Zn | Dosis actual de Zn (mg por toma) | Zn orina último valor | ALT(ULN) en el Dx | ALT(ULN) último valor | Cu orina (μg/24h) en el Dx | Cu orina (μg/24h)último valor |
| P1 | 26,3 | T y P | 0,8 | 25-25-25 | 249 | 221 (30) | 17 (31) | 254 | 67 |
| P2 | 8,7 | P | 6,3 | 25-25-25 | 569 | 11 (31) | 11 (55) | 50 | 26 |
| P3 | 28,3 | P | 2,4 | 50-0-50 | 5.022 | 22 (31) | 21 (55) | 65 | 48 |
| P4 | 2,8 | - | 2,8 | 25-25-25 | 5.627 | 67 (40) | 110 (55) | 59 | 69 |
| P5 | 3,7 | - | 3,7 | 25-0-25 | 3.496 | 61 (41) | 68 (55) | 91 | 43 |
| P6 | 18,3 | P y T | 6,5 | 50-0-50 | 2.433 | 28 (22) | 25 (55) | 31 | 47 |
| P7 | 26,3 | P | 1,3 | 25-25-25 | 2.882 | 496 (30) | 22 (55) | 130 | 34 |
| P8 | 3,3 | - | 3,3 | 50-50-50 | 10.642 | 70 (31) | 14 (31) | 194 | 86 |
| P9 | 7,6 | P | 6,3 | 50-50-50 | 2.100 | 238 (21) | 42 (55) | 88 | 56 |
| P10 | 2,3 | - | 2,3 | 50-50-50 | 1.879 | 191 (31) | 33 (31) | 95 | 32 |
| P11 | 5,3 | P | 3,3 | 50-50-50 | 3.830 | 113 (41) | 40 (41) | 118 | 57 |
| P12 | 5,5 | P y T | 3,4 | 50-50-50 | 6.187 | 33 (21) | 13 (31) | 476 | 43 |
| P13 | 28,3 | P | 1,0 | 50-0-50 | ND | 25 (31) | 12 (31) | 90 | 41 |
| P14 | 9,7 | - | 6,3 | 25-25-25 | 367 | 8 (31) | 7 (55) | 33 | 46 |
| P15 | 7,3 | P | 1,2 | 50-50-50 | 2.486 | 29 (22) | 36 (41) | 188 | 131 |
Dx: diagnóstico; P: penicilamina; Pac: paciente; T: trientina; ULN: límite superior normalidad.
En nuestro país existen varias series de pacientes de EW publicadas1,7–13. La mayor parte incluyen un número reducido de pacientes y no se acompañan de un estudio genético y/o de la aplicación de un sistema de puntuación («score») que confirme la enfermedad.
En nuestro estudio se describe la presentación clínica, diagnóstico (estudio genético, aplicación de sistemas de puntación) y evolución tras tratamiento de 15 pacientes con EW tratados en nuestro hospital. La discreta expresividad clínica y analítica de la EW en algunos de ellos, muestra la dificultad del diagnóstico de esta entidad en la práctica clínica diaria.
La EW es una enfermedad poco frecuente que debe ser descartada en presencia de una enfermedad hepática inexplicada y/o enfermedad neurológica-psiquiátrica sobre todo en pacientes jóvenes (menores de 50 años) y en sujetos con historia familiar de EW (familiares de primer grado)14.
La escasa sospecha de la EW por parte del clínico y su limitada sintomatología, a menudo inespecífica, hace que en muchos casos, esta entidad pase desapercibida y/o se diagnostique tardíamente. En nuestra cohorte, la presentación clínica «asintomática» ha sido la más frecuente siendo iniciado el proceso diagnóstico en su mayor parte tras estudio de una hipertransaminasemia. Además, en algunos casos, el diagnóstico de la enfermedad permitió atribuir a la propia EW sintomatología inespecífica trascendente que mejoró con el tratamiento: retraso estaturo-ponderal, dolor abdominal y astenia (P9), y alteraciones en el comportamiento-fracaso escolar (P10 y P11).
Por otra parte, la ausencia de un único marcador fiable que permita por sí solo confirmar o excluir la EW, hace que su diagnóstico constituya un reto en la práctica clínica diaria, sobre todo en aquellos pacientes asintomáticos con o sin antecedentes familiares de la enfermedad. Para facilitar el diagnóstico de la EW, diferentes guías clínicas6,15 recomiendan el estudio genético (mutaciones en el gen ATP7B) y la aplicación de «scores» o sistemas de puntuación, que combinan diferentes pruebas clínicas y analíticas3.
En la EW se han descrito más de 500 mutaciones distintas, algunas de ellas población-específicas, de las cuales 380 tienen un papel confirmado en la patogénesis de la enfermedad16. Por otro lado, más de un 50% de las mutaciones descritas en la EW son del tipo «missense» (con sentido erróneo) con dudas respecto a su implicación en el desarrollo real de la enfermedad17.
La mayoría de los pacientes con EW son heterocigotos compuestos de distintas mutaciones en el gen ATP7B. Además, los datos de la literatura no muestran una correlación entre el tipo de mutación ATP7B (genotipo) y el espectro clínico de la enfermedad (fenotipo)18. La presencia de mutaciones asociadas a la EW en ambos alelos (una mutación en homocigosis o 2 mutaciones distintas en heterocigosis) permite el diagnóstico de EW19. Por otra parte, la presencia de una única mutación en uno de los alelos no es concluyente de EW si no se acompaña de otros datos de la enfermedad o de antecedentes familiares de EW con dicha mutación. En nuestra cohorte, el estudio genético permitió el diagnóstico definitivo de EW en un familiar asintomático (P2) de una paciente con EW sintomática (P14). Por último, hasta en un 15% de los pacientes diagnosticados de la forma clásica de EW no se identifica ninguna mutación conocida en el gen ATP7B20. En nuestra serie, en un paciente con datos claros de EW no se identificaron alteraciones en la secuencia del gen (P10). Dos pacientes con datos clínicos y analíticos marcados de EW presentaron solo una mutación monoalélica (P1 y P7).
La mutación más frecuente en pacientes europeos con EW es la sustitución de una histidina por glutamato en la posición 1.069 (p.H1069Q), la cual se encuentra en el 40-60% de todos los casos21. En España, un estudio anterior demostró que la mutación más frecuente era la p.M645R10, presente en el 55% de las 40 familias estudiadas, aunque otros trabajos posteriores con series más cortas no han podido demostrar esta prevalencia7. En nuestro estudio la mutación p.M645R está presente en 8 de 11 (72%) de los sujetos índices de las familias (X2 p=0,29), pudiendo esta diferencia indicar un posible efecto fundador de esta mutación en nuestro medio. La frecuencia de las restantes mutaciones en la población nacional es menor, aunque en Canarias se ha descrito una alta frecuencia de la p.L708P22. Sin embargo, en nuestra serie, la mutación p.H1069Q, descrita como frecuente en otras poblaciones, solo está presente en 2 de 11 sujetos índices, uno de ellos con ascendencia centro-europea (P12).
Desde el año 2001 disponemos de un «score» diagnóstico de la EW, basado en datos clínicos, analíticos y genéticos3. Este sistema de puntuación, aunque escasamente validado en series amplias de enfermos adultos con EW, facilita el diagnóstico de la enfermedad incluso en pacientes asintomáticos, con escasos datos analíticos sugestivos de la enfermedad23. Tal es el caso de los pacientes P2 y P5, con estudio genético positivo y una ceruloplasmina discretamente disminuida, donde el «score» permitió el diagnóstico de la enfermedad y el establecimiento del tratamiento, en ausencia de realización de una biopsia hepática (P2) e incluso en presencia de una cuantificación de cobre intrahepático normal (P5). Ello remarca el papel relevante del estudio genético en este «score» diagnóstico de EW3,16,23.
La presencia de un Cu en orina >100μg/24h es un hallazgo frecuente en la EW, aunque valores entre 40-100μg/24h se pueden observar en pacientes pediátricos y en adultos presintómaticos con EW14,24. En nuestro estudio, 2 pacientes (13%) presentaron una excreción urinaria de Cu normal (<40μg/24h) (P6 y P14). La prueba de provocación con D-penicilamina se ha utilizado en el diagnóstico de la EW en población pediátrica25. Aunque está incluida en el «score» diagnóstico de la enfermedad, no se ha demostrado su utilidad en la población adulta6,24. En nuestra serie, debido al acceso del estudio genético, no se planteó la realización de la prueba de provocación con D-penicilamina para el diagnóstico de la EW.
Un 85% de nuestros pacientes tenían un contenido de Cu hepático mayor de 250μg/g t.s. Esta medida se considera como la mejor evidencia bioquímica para el diagnóstico de EW. Sin embargo, la presencia de un Cu intrahepático normal no excluye el diagnóstico de la enfermedad si el paciente presenta estudio genético compatible5,22,26.
El anillo de KF solo estuvo presente en 2 pacientes (13%), una con hipertransaminasemia asintomática (P6) y otra con cirrosis hepática-ascitis (P12). Ninguno de los casos con clínica neurológica o psiquiátrica presentaba anillo de KF (P13, P14 y P15). Clásicamente se ha considerado el anillo de KF como el sello clínico de la EW, estando presente en el 95% de los pacientes con síntomas neurológicos y alrededor del 50% de aquellos sin síntomas neurológicos6,21,24. Probablemente el alto número de pacientes que hemos diagnosticado en la fase precoz de la enfermedad haya influido en los pocos casos de anillo de KF observados.
Los tratamientos aprobados en la actualidad en la EW incluyen agentes quelantes del Cu (D-penicilamina y trientina) que favorecen su excreción urinaria y fármacos que inhiben su absorción intestinal (acetato de cinc).
En la actualidad y debido a su eficacia y mejor tolerancia, se recomienda el cinc como tratamiento de elección en sujetos con EW especialmente en pacientes asintomáticos y en aquellos con sintomatología neurológica, reservándose los agentes quelantes D-penicilinamina y trientina para el manejo inicial de los pacientes con enfermedad hepática sintomática donde el cinc podría ser menos seguro6,13,27. El tratamiento debe ser iniciado en el momento del diagnóstico, incluso en la fase presintomática, y mantenido de por vida6,24.
En nuestra serie, aunque inicialmente un porcentaje importante de pacientes fueron tratados con agentes quelantes, al final del estudio todos seguían tratamiento de mantenimiento con cinc. En los pacientes con afectación hepática, el tratamiento se asoció a una mejoría-normalización de la hipertransaminasemia y a una recuperación de la función hepática. Sin embargo, y probablemente debido a la instauración tardía del tratamiento, no se observaron cambios sustanciales en la evolución de los pacientes con sintomatología neurológica-psiquiátrica.
En conclusión, nuestra serie refleja la complejidad de la EW en la práctica clínica habitual, mostrando el papel relevante del estudio genético en el diagnóstico de la enfermedad. La identificación en nuestro medio de una mutación (p.M645R) en la mayoría de nuestros pacientes debe tenerse en cuenta en la estrategia para el análisis molecular del gen ATP7B en nuestra población28.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

