




El divertículo de Meckel constituye la anomalía congénita más frecuente del tubo digestivo (0,3-4%). Por su origen embrionario, la persistencia del conducto onfalomesentérico, este divertículo posee todas las capas propias de la pared intestinal, una irrigación propia e incluso su propio mesenterio, por lo que puede manifestarse clínicamente en el adulto mediante complicaciones propias de la mucosa intradiverticular, como la hemorragia secundaria a una secreción ácida por una mucosa gástrica ectópica, las alteraciones mecánicas (volvulación, intususpección o herniación) o, de forma infrecuente (0,5-3%), la malignización de alguna de sus capas con sarcomas (34%), tumores carcinoides (31,5%), adenocarcinomas (11,4%), leiomiomas (9,4%) y, excepcionalmente, tumores estromales gastrointestinales (GIST). A continuación exponemos un caso de GIST perforado sobre un divertículo de Meckel, circunstancia excepcional tanto por la concurrencia de ambas entidades en una misma paciente como por la forma de inicio.
OBSERVACIÓN CLÍNICAMujer de 46 años de edad, que acude al servicio de urgencias de nuestro hospital por presentar un cuadro de dolor abdominal de inicio brusco y carácter progresivo e intenso que comienza en el hipogastrio con generalización secundaria a todo el abdomen. Como antecedentes de interés, la paciente era alérgica a los salicilatos, había sido intervenida de una cesárea y apendicectomía en la juventud, padecía una anemia ferropénica de larga evolución, no estudiada, en tratamiento con sulfato ferroso, y una posible masa anexial derecha pendiente de valoración ambulatoria por su ginecólogo. El dolor abdominal se acompañaba de vómitos biliosos y mal estado general, con hipotensión leve y taquicardia; la paciente no presentaba fiebre, síndrome miccional, leucorrea ni metrorragia y la fecha de la última menstruación fue 15 días antes de su admisión en nuestro centro. En la exploración la paciente presentaba un abdomen con defensa generalizada mayor en el hipogastrio hacia la fosa ilíaca derecha, no tenía ruidos hidroaéreos y sí signos de irritación peritoneal. No se pudieron detectar masas a la palpación por la presencia de un abdomen en «tabla»; por otro lado, el tacto vaginal reveló la presencia de un importante dolor a la movilización del cuello uterino, sin constatarse leucorrea o alteraciones en esa localización. En el hemograma destacaba una anemia con hemoglobina de 10,7 g/dl, ya conocida y similar a rangos previos de la paciente, y una leucocitosis con neutrofilia propia del proceso agudo. El test de embarazo fue negativo.
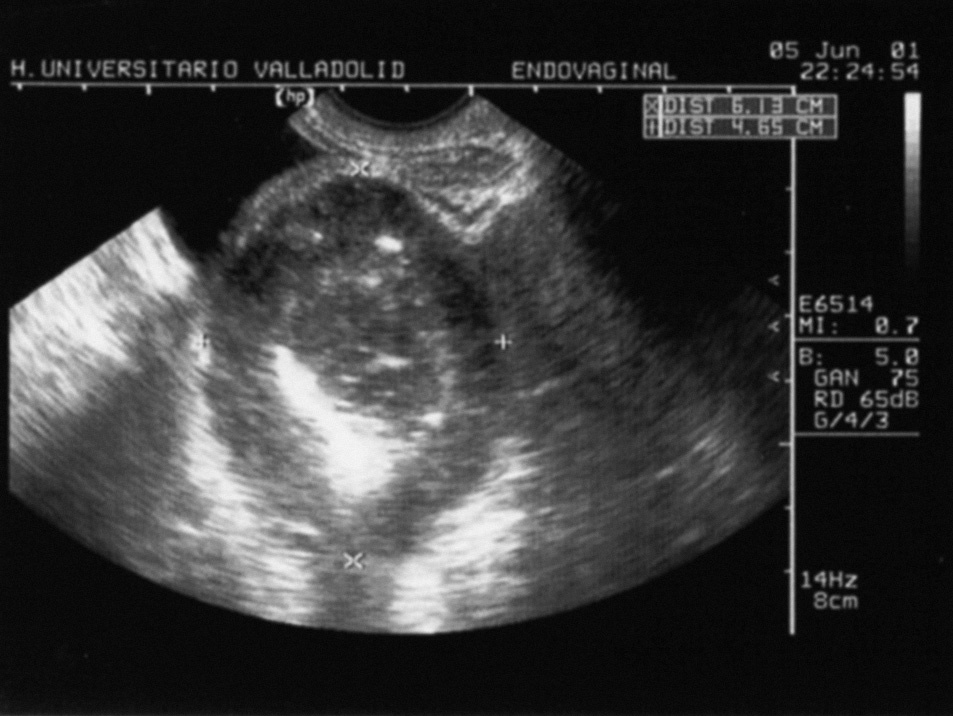
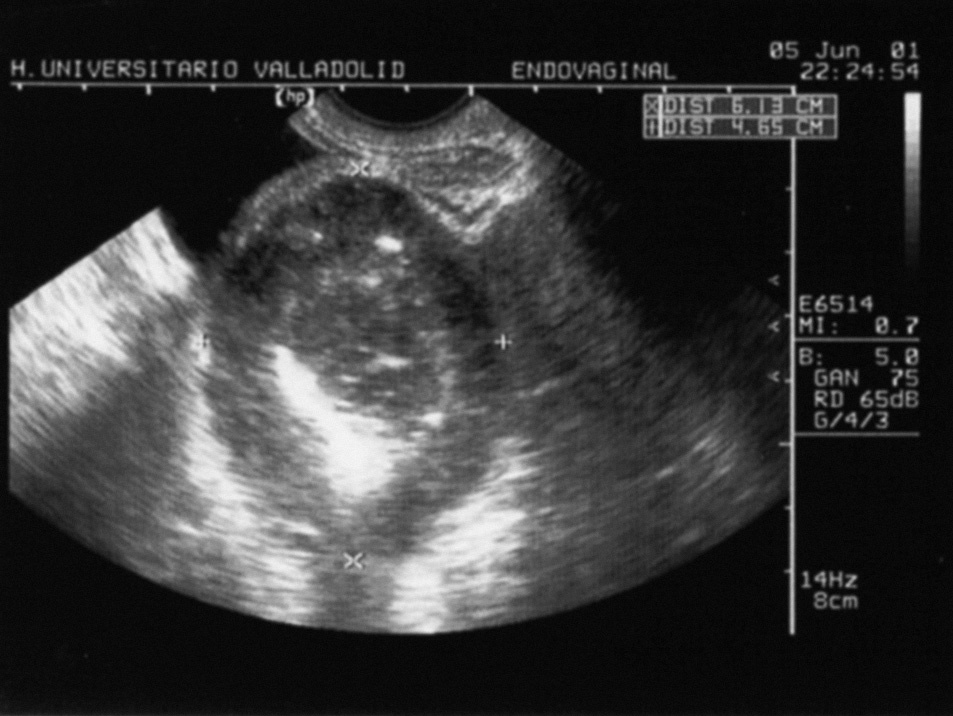
Fig. 1. Imagen de ecografía transvaginal urgente en la que se definió una «masa anexial mixta que parece depender de ovario derecho de 62 µ 48 mm, con signos de hemorragia intraquística y líquido libre en el espacio de Douglas».
Se realizó una ecografía transvaginal de urgencias con el resultado de un útero y un ovario izquierdo de características normales y una tumoración heteroecogénica de 62 µ 48 mm, que parecía depender del ovario derecho, con signos de posible hemorragia intraquística y líquido libre en el fondo del saco de Douglas (fig. 1). Ante tales hallazgos, la paciente fue intervenida con carácter urgente con el diagnóstico de una tumoración ovárica quística abierta al peritoneo. Se realizó una laparotomía media infraumbilical y se comprobó que la tumoración era dependiente del intestino delgado distal (próximo al íleon), situada en el borde antimesentérico, y dependía de un divertí-culo en la pared intestinal. La masa, de unos 7 cm de diámetro, presentaba un aspecto blanquecino, de superficie irregular, con una pared necrohemorrágica, elástica y trabeculada de unos 2 cm de grosor. Adyacente al divertículo, y posiblemente por el importante efecto traccional de la masa, se apreció una solución de continuidad de unos 2 cm de diámetro cubierta por fibrina, con una mucosa adyacente de carácter hemorrágico. Ambos ovarios eran normales y el útero sólo presentaba un carácter levemente miomatoso. Se realizó una resección con anastomosis terminoterminal primaria de la lesión y una exéresis en bloque del divertículo perforado y la tumoración, dejando 9 cm de seguridad en sentido proximal y distal a ésta. El análisis histológico reveló la presencia de una tumoración sobre un divertículo de Meckel con un patrón de crecimiento fascicular, constituida por células alargadas de hábito muscular liso con focos de fondo fibrilar tipo neuroide, cuyo origen se sitúa en una localización submuco-sa-muscular. La neoplasia presentaba una alta celularidad, un tamaño global de 7 cm, la presencia de necrosis central, aunque un bajo grado de pleomorfismo, una baja tasa mitótica (< 1 mitosis/10 campos de gran aumento [CGA]) y un índice proliferativo de ki-67 del 10%. Desde el punto de vista inmunohistoquímico, destacaba la positividad masiva de CD-117, la positividad alta de S-100 (100%) y de actina del 40%. Así pues, se definió como una tumoración GIST con diferenciación muscular lisa-neural y de carácter borderline. La evolución postoperatoria de la paciente fue excelente, y recibió el alta tras 6 días de ingreso hospitalario. Enviada a consultas externas de oncología, ante la inexistencia de una enfermedad residual, se decidió no iniciar tratamiento con quimioterapia o radioterapia adyuvante y establecer un seguimiento. Actualmente, 5 años después, se realiza un control ambulatorio clínico semestral y mediante tomografía computarizada (TC) abdominal, con una periodicidad anual, sin haberse observado ninguna recidiva local o a distancia. La anemia se corrigió en 3 meses sin necesidad de ferroterapia.
DISCUSIÓNLos GIST representan únicamente del 0,1-3% de todas las neoplasias del tracto digestivo, con una incidencia anual de 2 casos por 100.000 habitantes. De su origen mesodérmico similar al del resto de los sarcomas, de los que representan hasta el 20%1, las nuevas técnicas de tinción inmunohistoquímica desde el año 1998 nos han permitido separar estos tumores de otros similares desde el punto de vista histológico, como los leoimiomas, los leiomiosarcomas o los schwanomas, dada la presencia positiva del marcador CD 117 hasta en un 94% de los casos. Este marcador de membrana es compatible con la expresión celular de c-kit (4q11-21), protooncogén del que se deriva una glucoproteína con 5 dominios transmembrana tipo III de 145 kDa con actividad tirosincinasa y receptor habitual del SCF (stem cell factor), cuya acción es fundamental para la regulación correcta del ciclo celular; así pues, las distintas mutaciones que afectan a esa proteína serán las resultantes de la génesis tumoral directa de hasta el 90% de los GIST2. La mayor parte de estas neoplasias presentarán mutaciones en el dominio transmembrana en el exón 11 (80%), mientras que en la porción extramembrana (exón 9) o en el propio dominio tirosincinasa (exones 13 y 17) sólo se presentarán el 10-20% de las mutaciones; estos últimos, además, son más resistentes que el resto al tratamiento con mesilato de imatinib. Respecto a la patogenia, el resto de este tipo de tumores presenta mutaciones en otras proteínas, como la PDGFRA (factor de crecimiento derivado de plaquetas tipo alfa)3, similar en cuanto a tipo y funcionalidad a la anterior, y al gen de la neurofibromatosis tipo I4. Otras alteraciones relacionadas con las mutaciones de c-kit y PDGFRA se han investigado en pacientes con formas mutacionales familiares, y se han encontrado enfermedades, como la hiperpigmentación cutánea, la urticaria pigmentosa (V559A en c-kit) o la hiperplasia difusa de las células de Cajal en el plexo mioentérico5,6. Histológicamente, el origen del GIST reside en la capa submucosa de las células intersticiales de Cajal (CIC), involucradas en su función en la regulación motora del aparato digestivo7. El crecimiento de estas neoplasias es radial y puede originar una seudocápsula con tejidos vecinos normales no afectados en el 21% de los casos, infiltrativo en el 34%, o seudoexpansivo con formación de nódulos satélites en el 45%. Su aspecto anatomopatológico varía desde una diferenciación mioide a neural, con células fusiformes (70%), epitelioides (15%) o patrones mixtos (10%), y expresiones inmunohistoquímicas de S100 (neural), actina y desmina (mioide) o CD 34 y vimentina en los más indiferenciados8. La localización más frecuente de estas neoplasias es el estómago, con predominio de la región fúndica (52%), seguidos del intestino delgado (25%), ele colon (10%), el esófago (10%) y el mesenterio y el retroperitoneo (5%). No hay diferencias en la distribución entre sexos, pero sí es más frecuente su desarrollo entre la quinta y la sexta década de la vida. Clínicamente, se diagnostican con más frecuencia en el adulto, en torno a la quinta década de la vida, a través de la detección de una masa abdominal (38%), dolor abdominal inespecífico (40%) o sangrado gastrointestinal (30%). Otras complicaciones son más infrecuentes, como la perforación del tumor (8%). En cuanto al diagnóstico, cabe decir que en las pruebas de imagen los GIST aparecen como lesiones ocupantes de espacio intraabdominales heterogéneas, a menudo con zonas de necrosis parcheadas y de tamaño variable, con o sin afectación hepática secundaria en forma de metástasis9; endoscópicamente, aparecen como lesiones submucosas ulceradas o no, y las biopsias resultan a menudo negativas, dado el recubrimiento mucoso normal. La prueba aislada más rentable para el diagnóstico definitivo del GIST es la ecoendoscópica, con punción-aspira-ción con aguja fina (PAAF) asociada10; ésta permite el diagnóstico de extensión local, el tamaño, las características ecogénicas (factor pronóstico) y un diagnóstico histológico previo a la cirugía. La PAAF percutánea, en el caso de GIST potencialmente resecables, no se aconseja, dada la posibilidad de microsiembra en el trayecto de la aguja. La agresividad de estas neoplasias es variable, de carácter claramente maligno hasta en el 30% de los casos, e implica un mal pronóstico. Los factores relacionados con la agresividad son el tamaño mayor de 5 cm, un alto índice mitótico (> 5 mitosis por CGA), la existencia de metástasis o adenopatías al inicio, la recurrencia local o a distancia a menos de un año tras la resección completa, la necrosis, la alta celularidad, el pleomorfismo celular, la mutación en el exón 11 de c-kit y la existencia de mutaciones que promueven la resistencia a imatinib11,12. Pueden aparecer otros tumores hasta en el 5% de los pacientes con GIST; los más frecuentes son los de mama, los uterinos y los prostáticos. En cuanto a las opciones de tratamiento, se puede decir que la única curativa es, como en otros sarcomas, la resección «en bloque» con márgenes de seguridad adecuados13 (> 2-3 cm de márgenes sanos), dado el carácter infiltrativo de tales lesiones. La radioterapia no tiene un papel relevante. El único agente quimioterapeútico eficaz es el mesilato de imatinib (ST1571)14, fármaco que inhibe la actividad tirosincinasa tanto de c-kit como de PDGFRA, e impide la activación constitutiva en estos tumores; su papel ha sido validado para su uso en la leucemia mieloide crónica (LMC)15 y los GIST metastáticos o irresecables. El tratamiento estabiliza la enfermedad en el 30% de los casos y hay respuesta de regresión parcial en el 50%, entendiendo como tal una respuesta radiológica en la tomografía computarizada (TC) con un descenso de su diámetro mayor del 10% o un descenso de su densidad mayor al 15% de unidades Housnfield (HU), todo ello indicativo de necrosis y bloqueo de la actividad metabólica tumoral. El tratamiento con imatinib suele ser bien tolerado, aunque puede presentar efectos secundarios menores a las dosis empleadas habitualmente (400 mg/día), como edemas, fatiga, erupciones cutáneas, nauseas, vómitos, dolor abdominal, toxicidad medular (menos frecuente que en el uso para la LMC) o sangrado gastrointestinal secundario a necrosis parcial del tumor. El tratamiento, una vez instaurado, se mantiene de forma indefinida, y no hay evidencias en la literatura médica que apoyen su uso como agente adyuvante o neoadyuvante a la cirugía. En la pérdida de respuesta terapéutica al imatinib se han involucrado factores como la activación de dominios tirosincinasa alternativos, el aumento de receptores de membrana c-kit, y el más importante, la presencia de mutaciones de novo que escapen a la actuación del fármaco16. En estas situaciones los nuevos fármacos desempeñan un papel fundamental; a modo de ejemplo, el SU11248, un nuevo inhibidor de la tirosincinasa, ha demostrado un beneficio clínico relevante en estudios en fase III en pacientes con GIST resistente al imatinib17. Respecto al seguimiento de los pacientes con GIST, la prueba de imagen más empleada es la TC abdominal. Este método identifica la recidiva tanto local (lecho quirúrgico o implantes peritoneales) como a distancia, habitualmente en forma de metástasis hepáticas. La afectación ganglionar no es frecuente y la vía hematógena es la habitual para la diseminación tumoral; de hecho, en la cirugía la linfadenectomía regional sistemática no está estandarizada en los protocolos quirúrgicos. En cuanto a la frecuencia de las pruebas de imagen, no hay un consenso claro. Gómez-Senent et al18 aconsejan la realización de TC cada 3-4 meses hasta el tercer año, cada 6 meses hasta el quinto y posteriormente anual en los GIST de riesgo medio-alto; en los de bajo riesgo se pueden realizar revisiones cada 6 meses. El carácter altamente recurrente de estos tumores precisa un seguimiento a largo plazo, dado que se han reportado numerosas recidivas en períodos largos (10 años) con exéresis completa, aunque lo más habitual es que éstas se produzcan en los 2 primeros años. De hecho, Ng et al19 sugieren que sólo el 10% de los pacientes están libres de enfermedad a largo plazo. El divertículo de Meckel es la anomalía congénita más habitual del tracto gastrointestinal, cuya frecuencia oscila entre el 1 y el 3% de la población general20. Su origen radica en la persistencia del conducto onfalomesentérico, estructura que une el saco vitelino con el intestino en desarrollo; en condiciones normales se oblitera en la octava semana gestacional. El fallo en este proceso puede dar lugar a una fístula onfalomesentérica, un quiste entérico, una brida que unirá el intestino con el ombligo o, lo más frecuente, un divertículo de Meckel. El divertículo asentará en la cara antimesentérica y poseerá todas las capas propias de la pared intestinal, su propio meso y su vascularización propia procedente de la arteria mesentérica superior; su interior se revestirá de mucosa intestinal o ectópica, pancreática, colónica, hepática o, muy frecuentemente, gástrica. Su situación habitual es el yeyuno distal a menos de 100 cm de la válvula ileocecal y puede asociarse a otras malformaciones, como útero bicorne, páncreas anular o fisura palatina. Habitualmente es asintomático. La manifestación más común en niños menores de 5 años es la hemorragia digestiva baja, producida por una secreción ácida de la mucosa gástrica ectópica del divertículo que promueve la ulceración de la mucosa circundante. En la edad adulta se presenta a modo de complicaciones mecánicas: obstrucción, intususpección, volvulación, herniación, infección o torsión diverticular. La transformación neoplásica es poco habitual y los sarcomas y los carcinoides son los más frecuentes; se han comunicado GIST en casos aislados en la literatura médica21,22. El diagnóstico a menudo se realizará mediante laparotomía como un hallazgo incidental, si bien en el caso de alta sospecha, y si el divertículo presenta mucosa gástrica ectópica, la gammagrafía con pertenectato de tecnecio-99m puede revelar su presencia23. El tratamiento en el caso de complicaciones será quirúrgico24. El caso expuesto es un ejemplo de una rara asociación entre un divertículo de Meckel malignizado con un GIST en una mujer previamente asintomática. La presentación como abdomen agudo, la coexistencia en una misma paciente de dos entidades infrecuentes, el bajo índice de sospecha habitual en estas situaciones de una complicación de un divertículo de Meckel, así como la naturaleza tumoral de la lesión (GIST), conforman la particularidad de este caso.
Correspondencia: F. de la Morena López. Infanta María, 9, 3.º, 7. 28050. Madrid. España. Correo electrónico: felipe_de_la_morena@hotmail.com
Recibido el 22-3-2007; aceptado para su publicación el 14-5-2007.

