





La principal causa de falta de respuesta a la dieta sin gluten es la ingestión continuada y generalmente inadvertida de gluten. El diagnóstico de enfermedad celíaca refractaria se establece tras la exclusión de otras enfermedades, ante la persistencia de malabsorción y atrofia vellositaria. Comprende un heterogéneo grupo de afecciones, en general en pacientes adultos, y afortunadamente se manifiestan de forma infrecuente (< 5% de la población celíaca). Para su diagnóstico es fundamental la detección de alteraciones en la población linfocitaria intraepitelial. Ciertas alteraciones en estos linfocitos, como la ausencia de expresión de los receptores de superficie de la célula T (CD3 y CD8), son indicativos de una forma agresiva con potencial de transformación neoplásica (enfermedad celíaca refractaria tipo II). El tratamiento se basa en un adecuado soporte nutricional y en el empleo de corticoides y/o inmunosupresores (azatioprina e infliximab, principalmente). Ante un diagnóstico de enfermedad celíaca refractaria tipo II, el elevado riesgo de progresión a linfoma intestinal de células T obliga a emplear diferentes esquemas terapéuticos, en general más agresivos. Aunque actualmente ningún tratamiento ha demostrado claramente ser eficaz a largo plazo, la inmunoterapia con anti-CD52 o similares, y el trasplante autólogo de médula son dos opciones a tener en cuenta en enfermedad celíaca refractaria tipo II. Son prometedores los ensayos con anticuerpos que bloquean la secreción epitelial de interleucina 15, que es una molécula clave en la patogenia.
The main cause of lack of response to a gluten-free diet is continued, usually inadvertent, gluten intake. Diagnosis of refractory celiac disease is established on the basis of exclusion of other disorders, persistence of malabsorption and villous atrophy. Refractory celiac disease affects a heterogeneous group of patients, usually adults and, fortunately, is infrequent (< 5% of the population). Detection of alterations in the intraepithelial lymphocyte population is essential for diagnosis. Some alterations in these lymphocytes, such as the absence of T cell surface receptor expression (CD3 and CD), indicate an aggressive form of the disease with the potential for malignant transformation (type II refractory celiac disease).
Treatment is based on adequate nutritional support and on the use of corticosteroids and/or immunosuppressive agents (mainly azathioprine and infliximab). Because of the high risk of progression to intestinal T cell lymphoma, patients diagnosed with type 2 refractory disease require different –generally more aggressive– therapeutic strategies. At present, no treatment has been demonstrated to be effective in the long term, but two options that should be considered in type II disease are immunotherapy with anti-CD52 or similar agents, and autologous bone marrow transplantation. Trials with antibodies that block epithelial secretion of interleukin-15, a key molecule in the pathogenesis of the disease, are promising.
La enfermedad celíaca (EC) es una enteropatía inducida por el gluten de la dieta mediada por un mecanismo inmune en sujetos genéticamente predispuestos. La retirada del gluten se acompaña de la recuperación clínica e histológica en la mayoría de los pacientes. Un pequeño porcentaje de sujetos (< 5%) no responde a la dieta, por lo que presentan una enfermedad celíaca refractaria (ECR)1. La ECR es una entidad relativamente infrecuente, de aparición en la forma adulta de la EC, y puede cursar con una elevada morbimortalidad.
RESPUESTA A LA DIETA SIN GLUTENDías o semanas después del inicio de la dieta sin gluten (DSG), se observa una mejoría importante de la clínica. Las lesiones histológicas se recuperan más lentamente y, sobre todo en adultos, pueden tardar meses e incluso persistir en un 20–30% de los casos durante años en ausencia de sintomatología2,3.
Hasta un 30% de los pacientes inicialmente diagnosticados de EC pueden seguir presentando signos clínicos a pesar de realizar una correcta DSG. En estos casos, tras haber revisado el diagnóstico inicial, hay que descartar numerosas causas que pueden ocasionar la falta de respuesta a la dieta (tabla I).
Causas de no respuesta a la dieta sin gluten
| Incumplimiento de la dieta (latente o desconocido) |
| Intolerancia a otros alimentos (lactosa, fructosa) |
| Sobrecrecimiento bacteriano |
| Insuficiencia pancreática |
| Colitis microscópica o colitis colágena |
| Enfermedad inflamatoria intestinal |
| Esprue colágeno |
| Giardiasis |
| Yeyunitis ulcerativa |
| Enteropatía autoinmune |
| Linfoma intestinal |
| Otros tumores |
| Enfermedad celíaca refractaria |
La ingesta continuada de gluten, generalmente de una forma inadvertida y regular, es la principal causa de persistencia de la clínica. Hay que descartar los fármacos u otras sustancias que contengan gluten en forma de excipiente. La persistencia de títulos elevados de anticuerpos (antitransglutaminasa y antiendomisio) son un buen indicador de que persiste un contacto con el gluten4–6. No obstante, se ha descrito que estos anticuerpos podrían perder sensibilidad para detectar transgresiones dietéticas menores, tanto en niños como en adultos6,7. En general, debe realizarse un interrogatorio exhaustivo, junto con un diario de la dieta, y solicitar la ayuda de un dietista o nutricionista.
La intolerancia a otros alimentos, principalmente hidratos de carbono, suele acompañar a la EC, sobre todo al inicio8–10. La realización de tests basados en la medición de hidrógeno espirado puede ser útil para evaluar una malabsorción de hidratos de carbono y, a la vez, para descartar un sobrecrecimiento bacteriano intestinal. Tanto la intolerancia a los hidratos de carbono como el sobrecrecimiento bacteriano pueden ser responsables de la persistencia de la clínica tras excluir el gluten de la dieta11.
Asociado a la atrofia vellositaria se puede presentar insuficiencia pancreática exocrina tanto en niños como en adultos10,12,13. La determinación de quimiotripsina y elastasa en heces puede ayudar al diagnóstico y a establecer la indicación para iniciar suplementos enzimáticos. Estos pacientes deberán recibir especial atención con objeto de determinar si esa insuficiencia pancreática revierte tras la DSG o, por el contrario, permanece como insuficiencia primaria.
La colitis microscópica es una entidad que comparte la predisposición genética del antígeno de histocompatibilidad clase A (HLA) con la EC14. Por ello, ante un diagnóstico de colitis microscópica, deberá descartarse una EC. Asimismo, ante una EC que no responde a la DSG deberá investigarse la posibilidad de una colitis9,15.
Se ha descrito una mayor prevalencia de la enfermedad inflamatoria intestinal entre la población de enfermos con celiaquía que entre la población general16. Por tanto, se debe descartar especialmente cuando no hay respuesta a la DSG. La EC se asocia con frecuencia a manifestaciones de autoinmunidad17. De este modo se pueden observar yeyunitis ulcerativa18 o enteropatía autoinmune19 como causa de persistencia de síntomas.
Se debe descartar siempre la presencia de malignidad, especialmente el linfoma intestinal de células T como complicación de la EC. La pérdida ponderal, el dolor abdominal y la sudoración nocturna son síntomas frecuentes cuando está presente este tumor20. La videocápsula endoscópica21,22 y la enteroscopia de doble balón23 son las dos técnicas que en la actualidad han demostrado ser de gran ayuda para la localización de estos tumores.
DEFINICIÓN DE ENFERMEDAD CELÍACA REFRACTARIAFinalmente, una vez descartadas estas causas, la ECR será diagnosticada por exclusión. La ECR fue originalmente descrita por Trier et al24, en 1978, para definir a los pacientes con atrofia vellositaria y diarrea persistente sin respuesta a la DSG durante al menos 6 meses. Recientemente la Asociación Americana de Gastroenterología25 la ha definido como la persistencia de atrofia vellositaria y malabsorción clínica que no responde a la DSG. Esta situación puede aparecer inicialmente sin llegar a responder a la DSG desde su diagnóstico (primaria) o en pacientes ya diagnosticados de EC que después de un tiempo variable dejan de responder a la DSG (secundaria)26. Para algunos autores la ausencia de respuesta inicial al gluten haría sospechar que no es realmente una EC, y lo denominan esprue refractario no celíaco27. En general, cuando no hay una respuesta inicial a la DSG habría que revisar el diagnóstico de EC. La existencia de datos compatibles (serología típica, HLA DQ-2 o antecedentes familiares) apoyan el diagnóstico de ECR.
Su frecuencia se sitúa por debajo del 5% de los pacientes con EC. En la literatura médica los casos se agrupan en centros de referencia con series que no superan la veintena de casos28. Este hecho dificulta la creación de criterios de uniformidad para su diagnóstico y tratamiento.
PATOGENIA Y CLASIFICACIÓNEn los últimos años se ha progresado en el conocimiento de los mecanismos patogénicos implicados en el desarrollo de la EC. La respuesta inmune adaptativa frente al gluten en la lámina propia se ha descrito con profusión. Los linfocitos presentes en la lámina propia reaccionan frente a los péptidos de gliadina una vez deaminados por la enzima transglutaminasa tisular. La presentación de estos péptidos está mediada por DQ2 y DQ8. Una vez reconocidos los péptidos de gliadina por estos linfocitos T (CD4+), se activan y secretan interferón-γ, que desencadena la respuesta inflamatoria y se relaciona directamente con la atrofia vellositaria29.
Sin embargo, se ha avanzado menos en la explicación del aumento en los linfocitos intraepiteliales (LIE) que aparecen ya en las primeras fases de la enfermedad y que no disminuyen tras la DSG30. Estos linfocitos T difieren fenotípicamente de los presentes en la lámina propia, ya que en su mayor parte son CD8+ con un aumento en la expresión del receptor para el antígeno de tipo γδ31. Actualmente, están recibiendo especial atención, por su implicación en las principales complicaciones de la EC, la ECR y el linfoma de células T de tipo intestinal32,33. La interleucina 15 producida por los enterocitos en estrecho contacto con estos LIE, parece desempeñar un papel fundamental en la homeostasis de esta población linfocitaria y en su potencial transformación en la ECR y en el desarrollo de linfoma34,35.
En sujetos sanos y pacientes con una enfermedad celíaca no complicada, los LIE expresan en superficie el marcador CD103, que los diferencia de los linfocitos de la lámina propia. Además, mayoritariamente tienen un fenotipo de linfocito T CD3+ CD8+, y pueden expresar el receptor de célula T (TCR) αβ o γδ31. En función de las características de esta población de LIE, se diferencian dos tipos de ECR con diferente enfoque terapéutico y pronóstico32:
- –
ECR tipo I. La población de LIE presenta el fenotipo de marcadores de superficie similar a los pacientes con EC activa sin haber comenzado DSG. Además, cuando por técnicas de biología molecular se analiza el reordenamiento de los genes del receptor de la célula T (TCR), se observa que es policlonal.
- –
ECR tipo II. En este caso el fenotipo de los LIE se encuentra alterado, y constituye una población «aberrante». Esta población linfocitaria ha perdido los marcadores de superficie (CD3, CD8 y TCR), conservando el CD103 que la caracteriza como intraepitelial, así como la expresión de CD3 intracitoplasmático. Además, esta población presenta un reordenamiento oligoclonal o monoclonal del TCR. Debido a estas características, a este tipo II de la ECR también se le denomina «linfoma críptico intestinal de célula T», considerado como un linfoma T latente36.
La clínica de diarrea asociada a malabsorción es común a los dos tipos de ECR. El tipo I se suele presentar en pacientes más jóvenes y la clínica es menos patente. Con frecuencia se pueden asociar otros trastornos autoinmunes, infecciones o fenómenos tromboembólicos1. En el tipo II la edad media es mayor (50–60 años) y la clínica suele ser más manifiesta, con malabsorción grave y pérdida ponderal. En algunos pacientes se pueden presentar lesiones cutáneas, principalmente en las extremidades, similares al pioderma gangrenoso, y también infecciones o fiebre sin una causa determinada32.
Mediante endoscopia se puede observar una atrofia de los pliegues en el duodeno y también ulceraciones que pueden hacer sospechar una yeyunitis ulcerativa. Estas ulceraciones también se pueden apreciar en el estómago y el colon en la ECR tipo II37. Para poder visualizar todo el intestino delgado y descartar la presencia de lesiones en diferentes localizaciones, puede ser útil el empleo de la cápsula endoscópica22. Las lesiones visualizadas por la cápsula pueden ser categorizadas mediante biopsia tomada con enteroscopia de pulsión o de doble balón (alcanza tramos distales con mayor facilidad)38.
Las técnicas de imagen, en especial la tomografía computarizada, ayudan a descartar la presencia de tumores, principalmente el linfoma. En ocasiones se observa un aumento en el tamaño y el número de los ganglios mesentéricos sin presencia de linfoma.
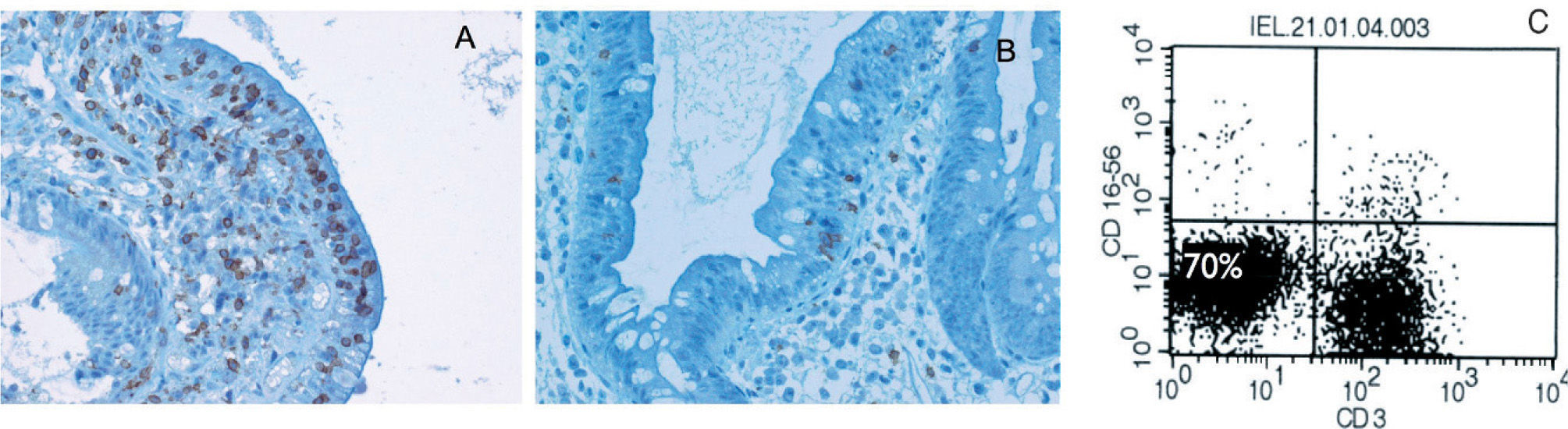
La histología presenta una clara atrofia vellositaria, similar a la que se puede apreciar en una EC que no ha comenzado con la DSG. Con las tinciones habituales no se puede diferenciar entre los dos tipos de ECR, por lo que es necesario realizar técnicas de inmunohistoquímica con tinciones frente a CD3 y CD8. Con ellas se observará que en ambos tipos de ECR hay un aumento de LIE que se tiñen con CD3 (se tiñe el citoplasma). Pero el primer dato que nos orienta hacia la ECR tipo II es que a diferencia del tipo I y de la EC sin refractariedad, estos LIE no se tiñen con CD839 (fig. 1).
 Inmunohistoquímica donde se observa un aumento de linfocitos, cuyo citoplasma se tiñe con el marcador CD3; sin embargo, esta población no se tiñe con el marcador para CD8 (B). C) Se observa esta población aberrante que no expresa el CD3 de superficie en casi el 70% de los linfocitos intraepiteliales, empleando citometría de flujo.")
Población linfocitaria aberrante presente en la enfermedad celíaca refractaria tipo II. A) Inmunohistoquímica donde se observa un aumento de linfocitos, cuyo citoplasma se tiñe con el marcador CD3; sin embargo, esta población no se tiñe con el marcador para CD8 (B). C) Se observa esta población aberrante que no expresa el CD3 de superficie en casi el 70% de los linfocitos intraepiteliales, empleando citometría de flujo.
Más útil e informativo es la realización de una citometría de flujo en muestras de biopsia para categorizar las poblaciones linfocitarias y cuantificar esa «población aberrante » de LIE (fig. 1). De este modo en la ECR tipo II se identifica una población mayoritaria que expresa CD103 en superficie (típico de los LIE y a diferencia de los linfocitos de la lámina propia), pero no expresa el CD3 de superficie (sí el CD3 intracitoplásmico que se observa en la inmunohistoquímica) ni el CD8 de superficie40.
Si nos encontramos ante un tipo II de ECR se debe buscar un posible reordenamiento clonal del TCR mediante técnicas moleculares. La presencia de oligoclonalidad o monoclonalidad está habitualmente asociada a la ECR tipo II, aunque no es imprescindible para el diagnóstico1.
La población de linfocitos aberrante presente en la ECR tipo II se puede encontrar no solamente en las biopsias de duodeno, sino también en el estómago, el colon y la sangre periférica37. Esto sugiere que la ECR tipo II es una enfermedad que no se limita al intestino delgado, sino que se expande al tracto gastrointestinal y puede diseminarse por vía hematógena.
EVOLUCIÓN Y PRONÓSTICOEn general, la ECR tiene un mal pronóstico, con una supervivencia menor del 50% a los 5 años de diagnóstico. Aunque la ECR es un grupo heterogéneo de entidades, la ECR tipo I podría representar un estadio más precoz de la enfermedad que la ECR tipo II, posiblemente con un evolución menos agresiva. El pronóstico va ligado a la presencia y el tamaño de la población de LIE aberrante, que condiciona el riesgo de desarrollo de linfoma intestinal41.
La presencia de clonalidad del TCR observada en la ECR tipo II se aprecia también en las muestras de linfoma intestinal. Esto sugiere una transformación de los linfocitos aberrantes que observamos en la ECR en un linfoma T de alto grado33.
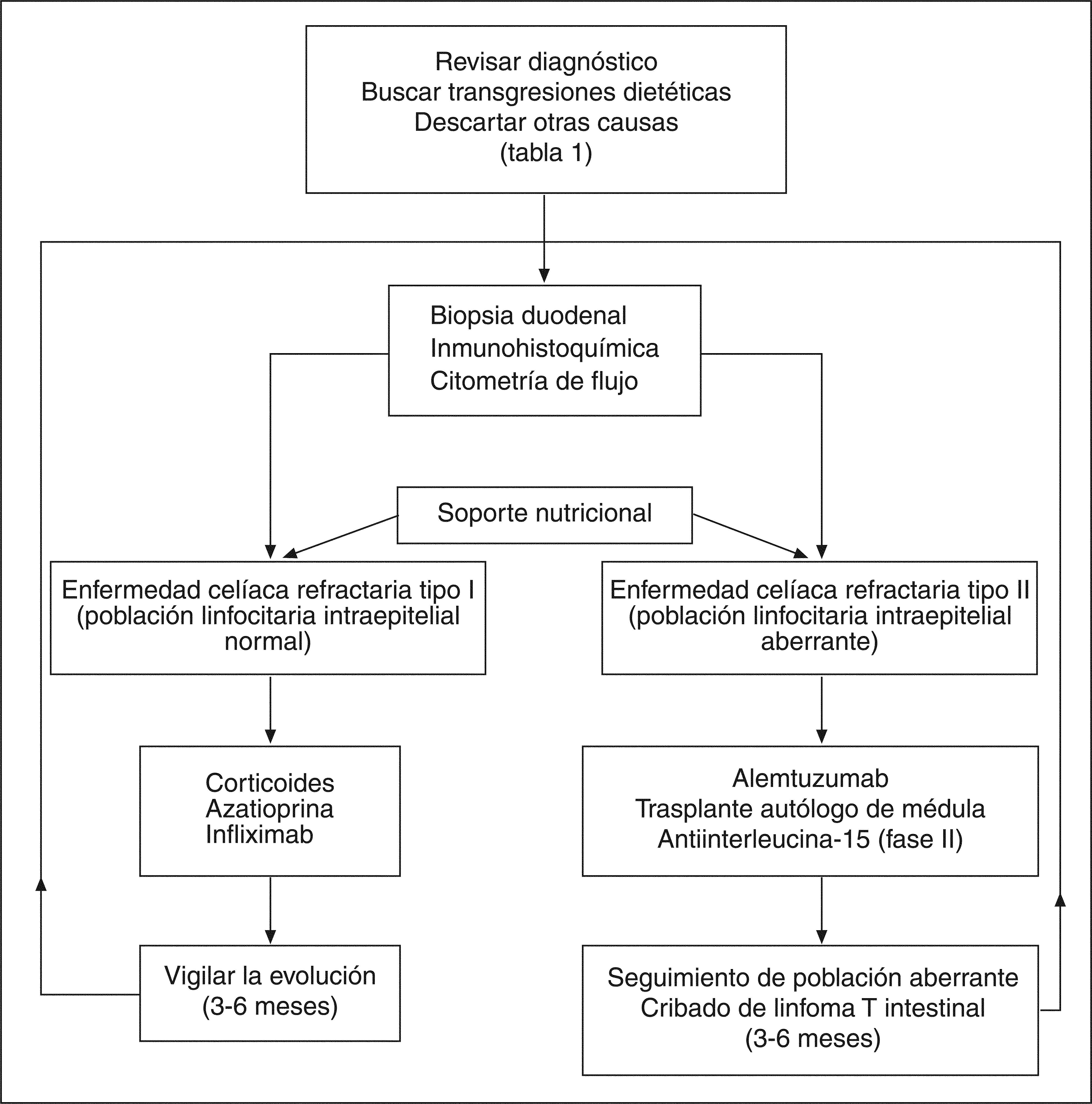
Actualmente no está claro cómo se debe hacer el seguimiento de los pacientes con ECR para una detección precoz de linfoma. La cápsula endoscópica puede poner de manifiesto lesiones tumorales precoces en el intestino delgado. También la tomografía por emisión de positrones podría diferenciar entre una ECR y un linfoma ya desarrollado42. En general, se debe realizar un estrecho seguimiento clínico y buscar la aparición de una neoplasia ante el deterioro del paciente o la aparición de síntomas de alarma. La toma de biopsias para el estudio histológico, inmunohistoquímico y citometría de flujo debería hacerse al menos cada 6 meses hasta la resolución de la refractariedad. Ante un tipo II habría que individualizar la toma de biopsias y acortar el intervalo (fig. 2).
TRATAMIENTO
El primer paso es un tratamiento de soporte principalmente nutricional, usando la vía parenteral si es necesario. Se deben corregir las alteraciones hidroelectrolíticas y el déficit de minerales (hierro, cinc, magnesio y calcio) y vitaminas (B12, ácido fólico, vitamina K y vitamina D). Por supuesto, se debe mantener una estricta dieta sin gluten.
Tratamiento de la enfermedad celíaca refractaria tipo IAdemás del soporte nutricional habitual, en este grupo de pacientes se ha ensayado una dieta elemental a base de aminoácidos. Los resultados mostraron una mejoría clínica e histológica junto con una disminución en la secreción mucosa de interleucina 15 e interferón γ en un grupo de pacientes con ECR tipo I43.
Los resultados observados con dieta elemental son a corto plazo, y se precisa avanzar en el escalón terapéutico. Aunque no hay estudios aleatorizados, los fármacos más empleados son los corticoides1. Se usan por vía intravenosa u oral según el grado de gravedad clínica, en una dosis de 1 mg/kg de prednisona o prednisolona. También se han empleados corticoides de acción local, como la budesonida con eficacia clínica similar44. En general, la respuesta clínica a los corticoides es buena a corto plazo, a pesar de que la mejoría histológica no se observa en un gran porcentaje de casos. Además, es frecuente la recidiva de la clínica al suspenderlos1.
Los casos con recaída tras suspender los corticoides o los de remisión clínica que han pasado una ECR tipo I, podrían ser candidatos a tratamiento inmunosupresor de larga duración. El fármaco más ensayado ha sido la azatioprina, con un elevado índice de respuesta clínica e histológica45,46. La dosis y la duración del tratamiento no han sido bien establecidas y, en general, se recomienda seguir la misma pauta que en la enfermedad inflamatoria intestinal. Con ciclosporina A, infliximab, tacrolimus y metotrexato se han obtenido resultados variables en casos clínicos aislados47,48. Quizá el infliximab ha sido el más evaluado y el que ha presentado mejores resultados en varios casos. Su uso se podría reservar para las situaciones de intolerancia a la azatioprina o falta de respuesta a ésta.
Tratamiento de la enfermedad celíaca refractaria tipo IINo hay un tratamiento establecido para esta forma agresiva de la ECR. No obstante, se admite que ante una población linfocitaria aberrante y clonal, el enfoque terapéutico debe ser más agresivo49. Aquí los corticoides o el infliximab pueden favorecer una mejoría clínica transitoria, pero sin ningún efecto sobre la proliferación clonal. Los inmunosupresores como la azatioprina pueden incluso favorecer la progresión a linfoma y no está recomendado su uso. La interleucina 10 humana recombinante se ha empleado con el objeto de inhibir la respuesta inmune de tipo Th1 frente a la gliadina. Sin embargo, no ha mostrado eficacia en una serie de 10 casos de ECR tipo II50.
Recientemente, se han ensayado agentes antineoplásicos, utilizados en el manejo de leucemias y linfomas. La cladribina (2-clorodesoxiadenosina) es un análogo sintético de la purina empleado en la leucemia de células peludas (tipo poco frecuente de linfoma T). Su empleo en una serie de casos de ECR tipo II ocasionó una mejoría clínica e histológica aunque con persistencia de la población linfocitaria aberrante y progresión a linfoma en el 40% de los casos51.
El alemtuzumab es un anticuerpo monoclonal anti-CD52, utilizado en el tratamiento de la leucemia linfocítica crónica. Su empleo en un caso de ECR tipo II originó una mejoría clínica e histológica junto con un descenso progresivo de la población de linfocitos aberrantes clonales y mantenimiento en remisión más de 18 meses41. La respuesta ha sido variable en otros casos, quizá asociado a diferentes estadios de la enfermedad.
El trasplante autólogo de médula después de quimioterapia intensiva se ha utilizado tanto en el linfoma establecido52 como en una serie de ECR tipo II, con buenos resultados clínicos, histológicos y de reducción de la población linfocitaria clonal53.
A pesar de todo, en la actualidad no se dispone de un tratamiento idóneo para esta población clonal presente en la ECR tipo II, por lo que se siguen buscando terapias innovadoras que actúen de modo más específico. En este sentido, el bloqueo de la interleucina 15 podría ser un enfoque prometedor. La producción de esta citocina está aumentada por el epitelio de los pacientes con ECR tipo II35. Además, se ha descrito que puede inducir linfoma cuando se sobreexpresa en ratones transgénicos54 y que dirige la expansión y la actividad de los LIE frente a los enterocitos34,35,55. De este modo, bloqueando su actividad se lograría no sólo eliminar la población de LIE aberrante, sino prevenir la destrucción del epitelio.
PUNTOS CLAVE PENDIENTES DE RESPUESTAAlgunas de las cuestiones y aspectos que deberán ser identificados en un futuro incluyen los siguientes:
- –
Factores que desencadenan la falta de respuesta a la DSG y la aparición de ECR.
- –
Mecanismos que inician la progresión a malignidad y diferencian los distintos estadios de la ECR.
- –
Realización de estudios multicéntricos que incluyan un elevado número de pacientes para poder extraer factores pronósticos y proponer un esquema terapéutico común.

