





RELEVANCIA CLINICA DE LA FIBROSIS INTESTINAL
La enfermedad de Crohn (EC) es una entidad relativamente prevalente en nuestro medio (100-150 pacientes por 100.000 habitantes, lo cual supone unos 50.000 pacientes en España y medio millón en Europa), y su incidencia ha aumentado muy notablemente en los últimos años. La EC se caracteriza por ser una enfermedad muy heterogénea, lo cual ha obligado a proponer diversas clasificaciones, basadas en el comportamiento clínico o el fenotipo de cada paciente. Las dos clasificaciones más recientemente descritas (Viena y Montreal) definen 3 fenotipos principales: inflamatorio, estenosante y fistulizante1,2. Los pacientes con EC estenosante se caracterizan por presentar una notable reducción de la luz que dificulta el tránsito intestinal (fig. 1). En estos pacientes los síntomas más frecuentes son el dolor abdominal, de características oclusivas, junto a la distensión abdominal, las náuseas y los vómitos, a menudo en ausencia de síntomas y parámetros analíticos sugestivos de actividad inflamatoria3,4.
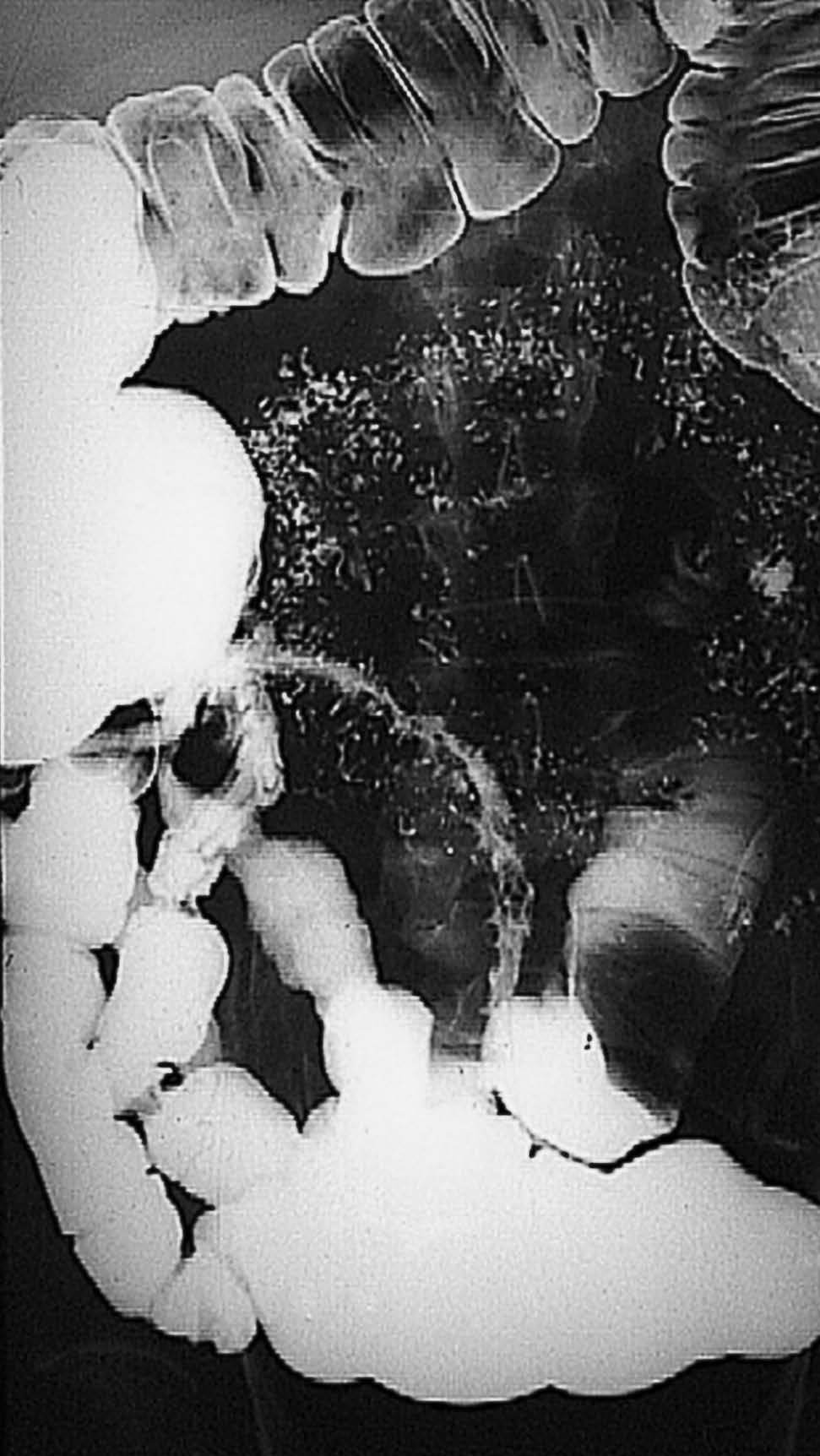
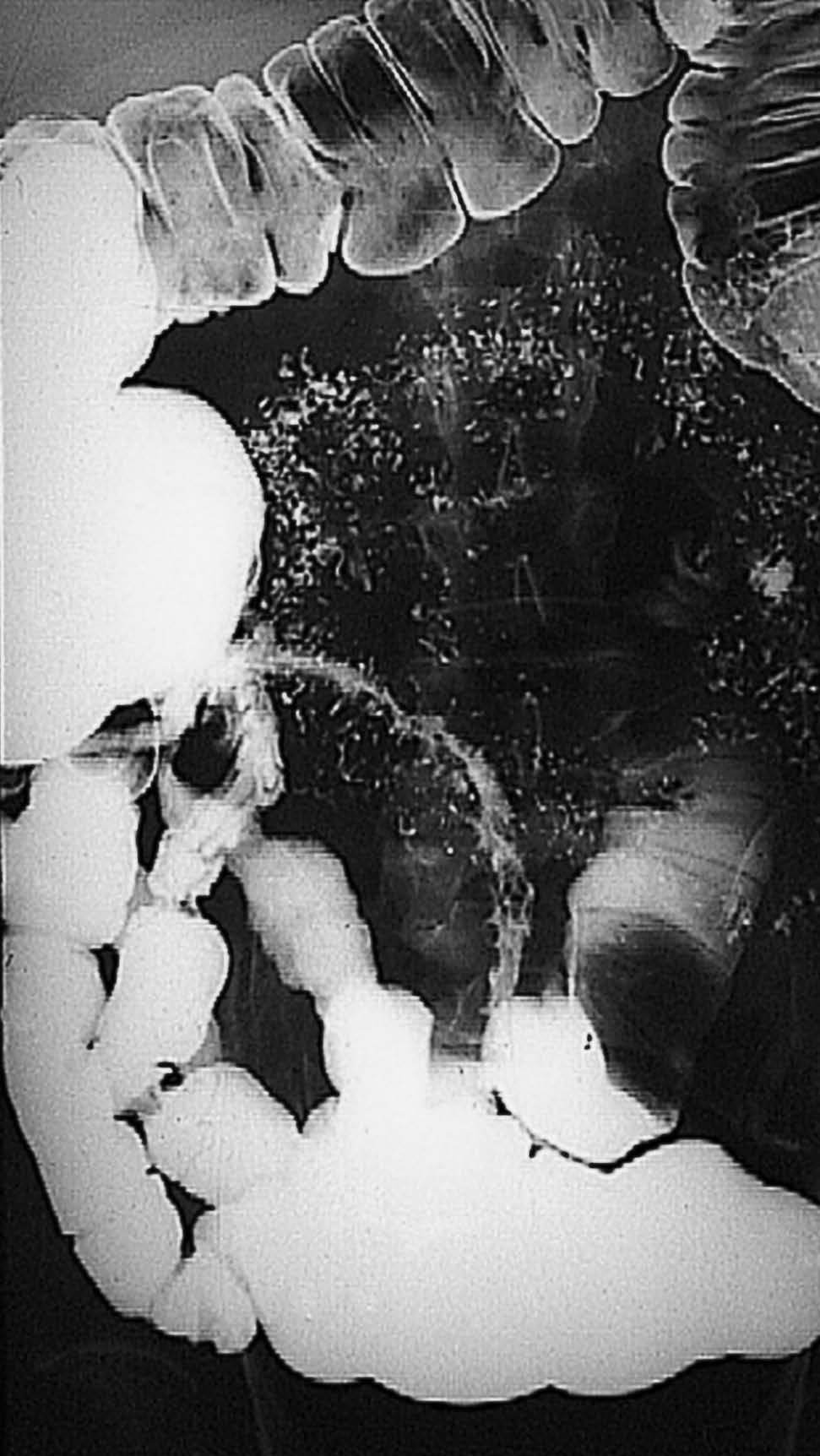
Fig. 1. Tránsito intestinal correspondiente a un paciente con enfermedad de Crohn de fenotipo estenosante. La imagen radiológica muestra una afección extensa del íleon terminal, con una clara reducción de su calibre, irregularidad de la mucosa y dilatación de la asa preestenótica.
Si bien sólo el 10% de los pacientes con EC presenta un fenotipo estenosante en el momento del diagnóstico de la enfermedad, el desarrollo posterior de estenosis es frecuente, y puede afectar en algún momento de su evolución a cerca del 50% de los pacientes con EC5. Es importante señalar que el desarrollo de un fenotipo estenosante condiciona notablemente la calidad de vida de los pacientes con EC, pues, además de los síntomas mencionados, es causa de repetidos ingresos hospitalarios, en ocasiones prolongados, debidos a episodios de oclusión o suboclusión intestinal. Al no haber ningún tratamiento médico específico para esta complicación, muchos de estos pacientes acabarán precisando tratamiento quirúrgico, que consiste en la resección del segmento intestinal afectado. Esta alternativa, además, tiene el inconveniente de no ser una solución definitiva para los pacientes con EC, pues la enfermedad reaparece en una gran proporción de éstos, pocos años después de la primera intervención, y el 40% de los casos precisa una segunda intervención quirúrgica6.
El desarrollo de estenosis en los pacientes con EC se debe a alteraciones en los mecanismos de reparación de las lesiones ulcerativas que aparecen en los brotes de actividad de esta enfermedad. La hipótesis más aceptada en la actualidad es que el subgrupo de pacientes que desarrollan una EC de fenotipo estenosante presenta una predisposición genética, que implica un proceso de reparación de la lesión intestinal anómalo, lo que conduce finalmente a una excesiva proliferación de fibroblastos y otras células mesenquimales y a un mayor depósito de colágeno, lo que acaba causando, en última instancia, la fibrosis intes tinal. No obstante, y a diferencia de la fibrosis hepática, pulmonar o cutánea, que han sido motivo de intensa investigación en los últimos años7,8, hay muy poca información sobre los mecanismos moleculares que regulan la fibrogénesis intestinal en la EC, algo realmente sorprendente si tenemos en cuenta su probada relevancia clínica.
FACTORES GENÉTICOS ASOCIADOS AL DESARROLLO DE ENFERMEDAD DE CROHN ESTENOSANTE
Es bien sabido que los factores genéticos desempeñan un papel importante en la fisiopatología de la EC. En los últimos años, numerosos estudios han intentado establecer la influencia que cada uno de dichos factores genéticos ejerce tanto en la susceptibilidad (riesgo de desarrollar la enfermedad), como en el fenotipo o curso clínico seguido por la EC. El factor genético asociado de forma más constante a un fenotipo estenosante de la EC es el NOD2/CARD156,9-11, cuyas tres variantes principales (SNP8, SNP12 y SNP13) son, además, el principal factor de susceptibilidad para desarrollar la EC. En ese sentido, resulta muy útil el metaanálisis publicado por Economou et al12 en 2004, en el cual se incluyeron datos procedentes de 42 estudios que habían evaluado la presencia de las tres principales variantes del gen NOD2/CARD15 en diferentes cohortes de pacientes con EC. Este metaanálisis demuestra que las variantes del gen NOD2 condicionan un moderado incremento del riesgo de presentar un fenotipo estenosante, cuya odds ratio es de 1,94 (intervalo de confianza [IC] del 95%, 1,61-2,34). Más recientemente, también se ha descrito la asociación de un polimorfismo (T280M) en el receptor de fractalkine, una quemoquina implicada en los fenómenos de quemotaxis y de adhesión leucocitaria al endotelio, a un mayor riesgo de presentar EC estenosante13.
SINTESIS DE COLAGENO Y OTROS COMPONENTES DE LA MATRIZ EXTRACELULAR
Diversos estudios realizados en las décadas de los ochenta y noventa utilizaron técnicas de inmunohistoquímica, hibridación in situ y northern blot para estudiar los tipos celulares presentes en la pared intestinal, así como para cuantificar el depósito de colágeno y de otros componentes de la matriz extracelular, en muestras de intestino de pacientes afectados de EC estenosante, EC no estenosante, colitis ulcerosa (CU) y en sujetos sanos. Graham et al14 demostraron que en el intestino de los pacientes con EC estenosante hay un mayor número de células musculares lisas, tanto en la capa muscularis mucosa como en la muscularis propia; asimismo, se produce una mayor síntesis y depósito de colágeno y se detecta también un aumento relativo del colágeno tipo V. Otros autores15, en cambio, al comparar los hallazgos obtenidos en los pacientes con EC y CU observaron que en el intestino de los pacientes con EC hay una mayor expresión de procolágeno I, III y IV que en el de los pacientes con CU. Más recientemente, se ha descrito la presencia de grandes acumulaciones de mastocitos en las estenosis intestinales. Los mastocitos se concentran en la capa muscular del intestino y su localización coincide con el depósito de laminina, pero no con el de otros componentes de la matriz extracelular, como fibronectina o vitronectina16. Esta observación sugiere que los mastocitos participan en la fibrogénesis intestinal, activando células mesenquimales de la capa muscular, que serían las responsables de la síntesis anómala de laminina. La presencia de unos valores altos de laminina y bajos de colágeno IV en el plasma de los pacientes afectados de EC y CU17 apoya esta hipótesis.
FACTORES PROFIBROGÉNICOS INTESTINALES
Una vez descubierta la presencia de alteraciones en la síntesis de colágeno y de otros componentes de la matriz extracelular en los pacientes con EC, el foco de atención de la mayoría de los trabajos se centró en el estudio de diversos factores profibrogénicos o antifibrogénicos, como posible explicación a las alteraciones observadas. Dammeier et al18 hallaron un notable incremento del ARN mensajero del connective tissue growth factor (CTGF) en el intestino de pacientes con EC o CU, respecto a muestras de intestino sano. Además, el mismo trabajo demostró que la expresión de dicho factor era máxima en zonas de fibrosis o inflamación muy grave, y también que había una estrecha correlación entre la expresión de CTGF y la de transforming growth factor (TGF) ß1 (su inductor), colágeno I y fibronectina. En otro estudio similar, Di Mola et al19 hallaron una expresión de CTGF 5 veces mayor en la EC que en la CU o en muestras de colon sano, utilizando hibridación in situ y northern blot.
Además del CTGF, dos de los factores con mayor capacidad para inducir fibrosis en diversos tejidos son el TGF-ß y el insulin-like growth factor (IGF) 1. En un interesante estudio, Lawrance et al20 demostraron que la localización de la inflamación intestinal (limitada a la lámina propia en la CU y transmural en la EC) es la que determina el incremento de la expresión de TGF-ß y IGF-1 en el intestino, el cual origina finalmente el depósito anómalo de colágeno, con un incremento de la ratio colágeno III/colágeno, y el desarrollo de fibrosis. En otras palabras, según estos autores sería únicamente la localización transmural de la inflamación, y no una alteración específica de la EC, lo que explicaría la tendencia a desarrollar estenosis intestinal transmural en la EC y no en la CU.
La presencia de una expresión aumentada de IGF-1 y de su receptor en los pacientes con EC se ha confirmado en otros estudios21,22, en los que se ha observado también un mayor número de fibroblastos y miofibroblastos, así como una mayor síntesis de procolágeno *1.
METALOPROTEINASAS Y DEGRADACION DE LA MATRIZ EXTRACELULAR
En cualquier tejido se produce de forma fisiológica un equilibrio entre la síntesis y la degradación de colágeno y de otros componentes de la matriz extracelular. Las metaloproteinasas son un grupo numeroso de enzimas implicadas en este último proceso, de forma que una alteración en su función (o en la de las moléculas que regulan su actividad) puede ser causa de fibrogénesis patológica.
En un primer estudio, Bailey et al23 hallaron tanto un aumento del número de células inflamatorias positivas para gelatinasa B como una mayor expresión extracelular de stromelysin, limitada a áreas de fibrosis intestinal en los pacientes con EC. Resultados parecidos se obtuvieron en un estudio más reciente, realizado en pacientes de edad pediátrica afectados de EC y CU. De nuevo, se observó un incremento de la expresión de la metaloproteinasa stromelysin, tanto en la EC como en la CU, respecto a los controles sanos. En cambio, no hubo diferencias en la expresión del tissue inhibitor of metalloproteinases (TIMP) 1 entre los 3 grupos24. Es posible que el incremento en la expresión de metaloproteinasas descrito en los dos trabajos anteriores no sea exclusivo de la enfermedad inflamatoria intestinal, sino común a cualquier otra entidad que curse con ulceraciones del intestino. Así lo sugieren los resultados de un estudio en el que, además de EC y CU, se incluyó a pacientes afectados de colitis isquémica. En este estudio se analizó la expresión de diferentes metaloproteinasas y sus inhibidores, mediantes hibridación in situ e inmunohistoquímica, y se concluyó que hay un patrón de sobreexpresión de diversos factores (colagenasa 3, stromelysin 2 y TIMP 3) común a las 3 entidades que cursan con ulceración colónica (EC, CU y colitis isquémica), que no se observa en el colon sano25.
TÉCNICAS DE AISLAMIENTO DE FIBROBLASTOS INTESTINALES
Es bien sabido que diversas células mesenquimales, como los fibroblastos, los miofibroblastos y las células musculares lisas, desempeñan un papel fundamental en el desarrollo de la fibrosis en diferentes tejidos. En cambio, es todavía motivo de controversia si la causa inicial de la fibrosis reside en alteraciones primarias de dichas células o bien si éstas sólo actúan como efectoras, respondiendo a señales que tienen lugar con anterioridad y, probablemente, relacionadas con el fenómeno inflamatorio. Para aclarar este punto, en el caso de la fibrogénesis intestinal asociada a la EC se han desarrollado en los últimos años diversas técnicas que permiten obtener fibroblastos y otras células mesenquimales a partir de muestras de intestino, para ser luego estudiadas in vitro, desde un punto de vista tanto morfológico y funcional.
Hay dos tipos de técnicas para obtener fibroblastos a partir de muestras de intestino: a) técnicas basadas en el sobrecrecimiento (outgrowth), que consisten en mantener un fragmento de mucosa intestinal en condiciones de cultivo favorables, de forma que, pasado un tiempo, los fibroblastos son los únicos tipos celulares capaces de adherirse al plástico y proliferar26,27, y b) técnicas basadas en la digestión enzimática del tejido, mediante las cuales se realiza un proceso de triturado mecánico y digestión enzimática de la mucosa intestinal, como paso previo al cultivo descrito anteriormente28.
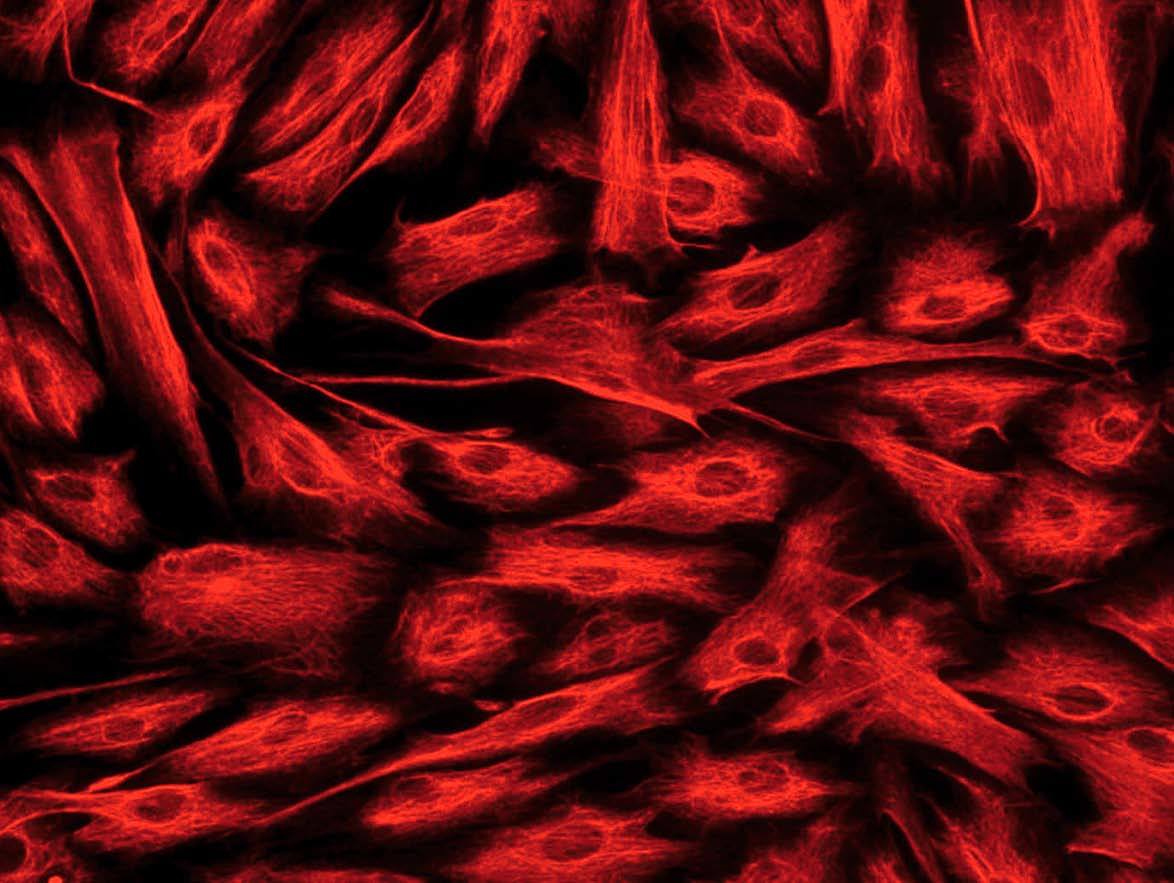
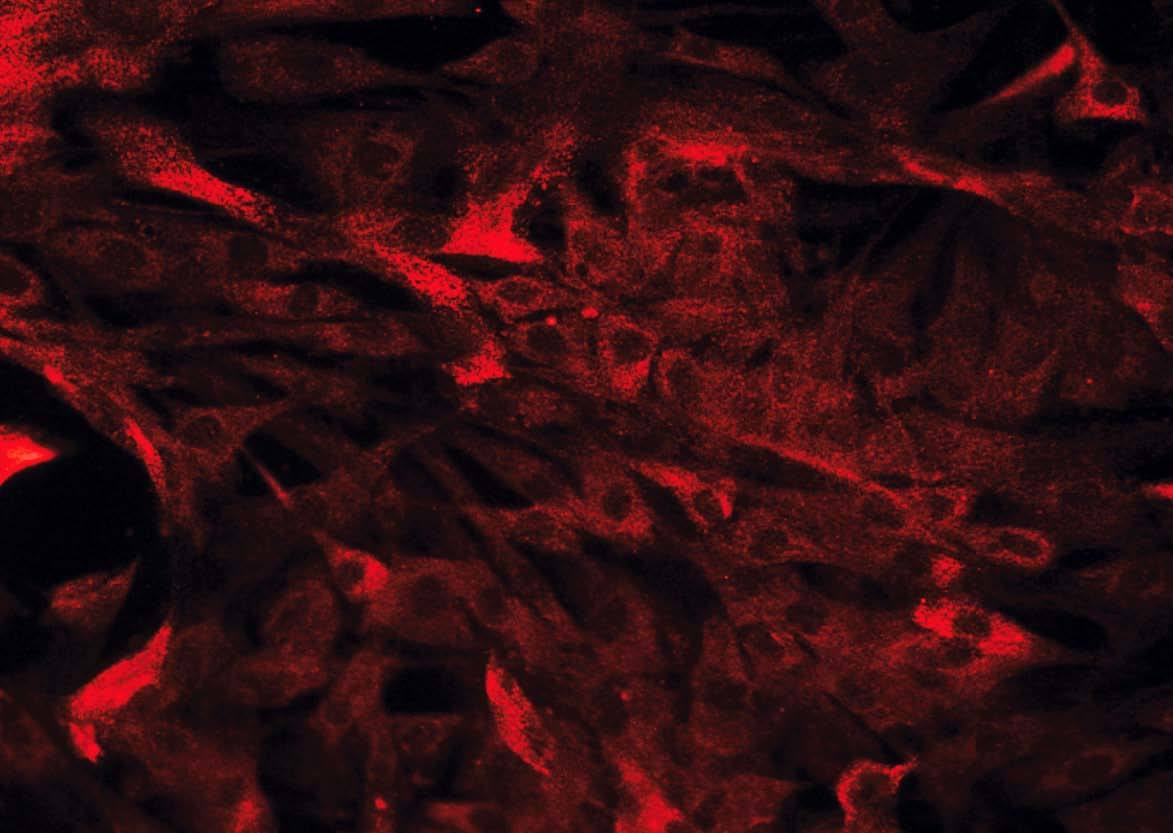
Hay diversos tipos de células mesenquimales que pueden aislarse de la pared intestinal. Si bien a menudo se hace referencia a ellas, de forma genérica, como «fibroblastos intestinales» o «células mesenquimales del intestino», pueden diferenciarse 3 tipos celulares distintos mediante inmunohistoquímica: a) fibroblastos, que son vimentina (+), *-actina () y desmina (); b) miofibroblastos, que se diferencian de los anteriores por ser *-actina (+), lo cual les confiere capacidad contráctil, y c) células musculares lisas, que son positivas para los 3 marcadores mencionados y suponen sólo un pequeño porcentaje del total de células aisladas20,22 (fig. 2).

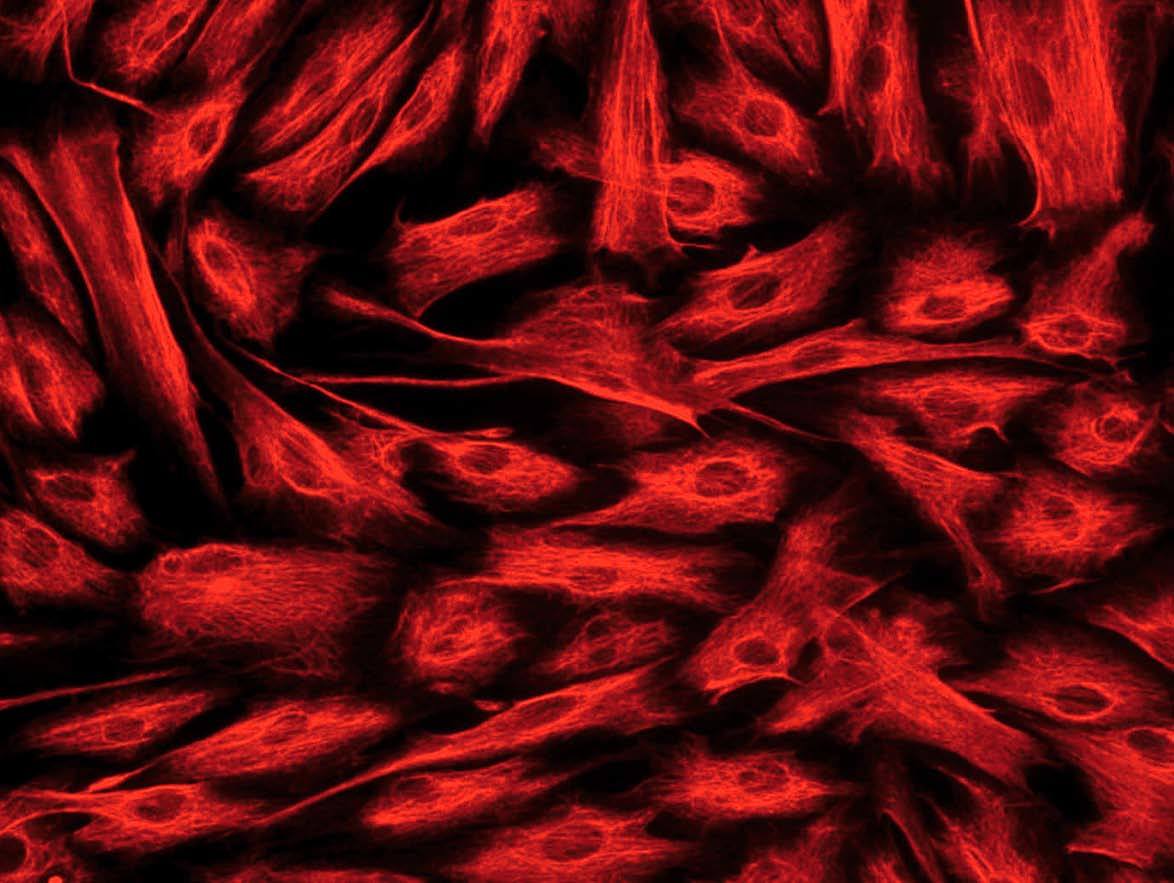
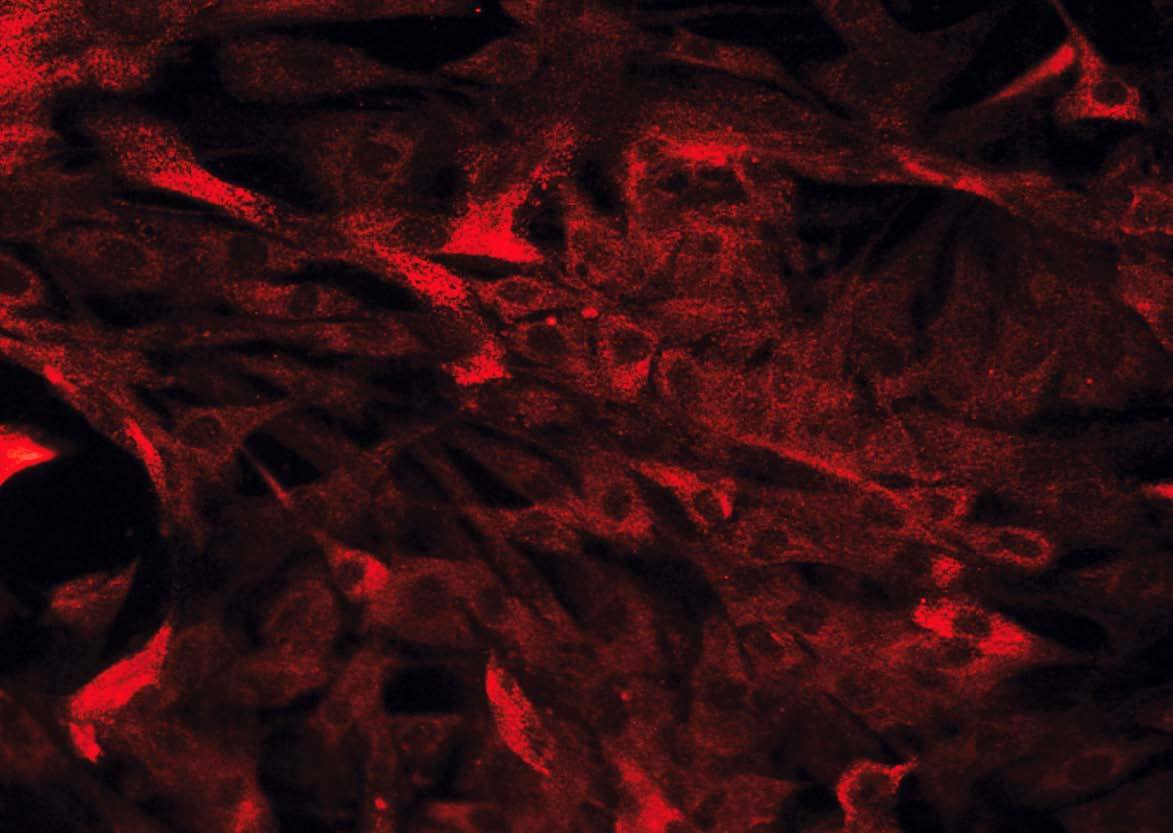
Fig. 2. Estudio inmunohistoquímico de un cultivo de fibroblastos intestinales, procedentes de un paciente con enfermedad de Crohn. A) La totalidad de las células presentan una intensa expresión de vimentina en su citoplasma. B) La tinción mediante *-actina permite diferenciar una proporción de células que expresan este marcador (miofibroblastos), mientras que la mayoría de las células son negativas para*-actina (fibroblastos). C) Tan sólo alguna célula aislada presenta positividad para desmina en este tipo de cultivos (células musculares lisas).
Las técnicas para aislar fibroblastos intestinales no proporcionan un aislamiento «puro», sino una mezcla de estos 3 tipos celulares. En este sentido, Strong et al29 demostraron que las técnicas que incluyen digestión enzimática de la mucosa proporcionan un mayor porcentaje de miofibroblastos que las técnicas de sobrecrecimiento. También se debe tener en cuenta que dicho fenotipo no es algo fijo en el tiempo, sino que puede producirse una transformación de un tipo celular a otro, en función de diferentes estímulos. Así, el TGF-ß induce la transformación hacia un fenotipo tipo miofibroblasto30, igual que cuando las células son cultivadas a baja densidad31, mientras que el IGF-1 induce la transformación de miofibroblastos en fibroblastos30.
ESTUDIOS MORFOLOGICOS Y FUNCIONALES EN FIBROBLASTOS INTESTINALES AISLADOS
Diversos estudios han demostrado que los fibroblastos intestinales procedentes de pacientes con EC poseen una serie de características funcionales específicas. Este hecho apoyaría la teoría que sostiene que hay una serie de alteraciones primarias en los fibroblastos intestinales como responsables, al menos en parte, de la fibrogénesis anómala que tiene lugar en la EC. En uno de los primeros estudios, Stallmach et al32 demostraron que los fibroblastos obtenidos de zonas estenóticas del intestino de pacientes con EC sintetizan una mayor cantidad de colágeno, especialmente de tipo III, y son más sensibles a la acción estimuladora del TGF-ß1. Dos estudios28,33 coinciden en señalar que los fibroblastos intestinales procedentes de pacientes con enfermedad inflamatoria intestinal (tanto EC como CU) proliferan de forma más rápida, tanto basalmente como en respuesta a diferentes estímulos, que los fibroblastos obtenidos a partir de muestras de colon sano. En uno de estos estudios33 la capacidad de proliferación fue mayor en la EC estenosante que en la CU, mientras que en el otro estudio28 no se observaron diferencias entre las dos enfermedades. También se han observado diferencias en relación con la capacidad de activación de los fibroblastos. Vogel et al27 demostraron que los fibroblastos intestinales procedentes de pacientes con EC y CU poseen una expresión mucho mayor de la molécula coestimuladora CD40. Al ser expuestos al ligando de esta molécula (CD40L), se produce una gran activación de los fibroblastos, demostrada por el incremento de diversas moléculas de adhesión en su superficie, como ICAM-1 y VCAM-1, la secreción de interleucina (IL) 8 y la capacidad de unirse a linfocitos. Finalmente, los fibroblastos procedentes de pacientes con EC estenosante expresan más TGF-ß2, menos TGF-ß3 y menos TIMP-1 que los fibroblastos obtenidos a partir de muestras de colon sano33,34.
MODELOS EXPERIMENTALES DE FIBROSIS INTESTINAL
A diferencia de lo que ocurre en la colitis experimental, en la que disponemos de un gran número de modelos animales diferentes, hay pocos modelos experimentales específicamente diseñados para reproducir la fibrosis intestinal. Este hecho ha sido una de las principales limitaciones para el desarrollo de nuevas estrategias de tratamiento antifibrogénico en el intestino. En el año 2003, Lawrance et al35 describieron un modelo de fibrosis intestinal en el ratón, inducido por la administración intracolónica repetida y en dosis ascendentes del hapteno TNBS. Los autores demostraron el desarrollo de un alto grado de fibrosis intestinal, de forma prácticamente constante, tanto en ratones CD1 como Balbc. Dos años más tarde, Vallance et al36 decribieron un nuevo modelo, basado en la transfección de TGF-ß 1 al epitelio colónico, mediante un vector administrado en forma de enema. Una vez demostrada la eficacia de la transfección en el epitelio colónico, los autores observaron que a dicha intervención le sigue la aparición de células con fenotipo miofibroblasto y un mayor depósito de colágeno, lo cual conduce a una intensa fibrosis intestinal y, a menudo, a la oclusión intestinal. Además de proporcionar un nuevo modelo experimental, este trabajo demuestra, una vez más, el papel clave que desempeña el TGF-ß en la fibrogénesis intestinal.
A diferencia de los modelos previamente mencionados, específicamente diseñados para estudiar la fibrosis intestinal, debe de tenerse en cuenta que en la mayoría de los numerosos modelos de colitis experimental actualmente utilizados (como la colitis inducida por TNBS o por peptidoglicano-polisacárido en la rata), además de las lesiones de tipo inflamatorio hay también un componente, más o menos intenso, de fibrosis intestinal.
TERAPIA ANTIFIBROGÉNICA INTESTINAL
Son relativamente pocos los estudios que han evaluado las posibles estrategias para prevenir o tratar la fibrosis intestinal. En el primero de ellos, Lawrance et al35 lograron atenuar de forma notable el desarrollo de fibrosis intestinal inducida por TNBS, especialmente cuando los animales fueron pretratados con un oligonucleótido antisentido para la subunidad p65 del factor de transcripción NF-*B, mientras que el efecto fue algo menor cuando el tratamiento con el oligonucleótido se realizó una vez ya establecida la fibrosis intestinal. En un segundo estudio se ensayó el tratamiento con captopril, un inhibidor de la enzima de conversión de la angiotensina, en la colitis inducida por TNBS en la rata. La elección de este fármaco se basa en el hecho de que la angiotensina II ha demostrado ser un factor clave en la fibrogénesis hepática, pulmonar y renal. El pretratamiento con captopril redujo significativamente la fibrosis intestinal que de forma característica presentan los animales 3 semanas después de la administración intracolónica de TNBS. En este estudio se demostró también una clara reducción del TGF-ß 1 colónico, lo cual sugiere que el efecto antifibrogénico logrado por el captopril podría estar mediado por este factor37.
Finalmente, en un estudio más reciente, Theiss et al38 demostraron que la administración de hormona del crecimiento tiene un efecto antifibrogénico moderado en el modelo de colitis experimental inducido por la injección subserosa de peptidoglicano-polisacárido en la rata, sin que se observe, en cambio, ningún efecto antiinflamatorio.
CONCLUSIONES
A pesar de su relevancia clínica, los mecanismos que regulan la fibrogénesis intestinal son todavía muy poco conocidos. Se ha descrito una serie de alteraciones, tanto en el tejido intestinal como en los fibroblastos aislados, que parecen contribuir a la fibrosis del intestino. No obstante, serán necesarios muchos más estudios para comprender mejor la fisiopatología de la fibrosis intestinal y desarrollar estrategias de prevención y tratamiento de esta complicación.

