




Nonalcoholic fatty liver disease (NAFLD) is the most common chronic liver disease in the world. Whereas insulin resistance and obesity are considered major risk factors for the development and progression of NAFLD, the genetic underpinnings are unclear. Before 2008, candidate gene studies based on prior knowledge of pathophysiology of fatty liver yielded conflicting results. In 2008, Romeo et al. published the first genome wide association study and reported the strongest genetic signal for the presence of fatty liver (PNPLA3, patatin-like phospholipase domain containing 3; rs738409). Since then, two additional genome wide scans were published and identified 9 additional genetic variants. Whereas these results shed light into the understanding of the genetics of NAFLD, most of associations have not been replicated in independent samples and, therefore, remain undetermined the significance of these findings. This review aims to summarize the understanding of genetic epidemiology of NAFLD and highlights the gaps in knowledge.
La enfermedad hepática no alcohólica (EHNA) es la enfermedad crónica del hígado más común en el mundo. Si bien se considera que la resistencia a la insulina y la obesidad son factores de riesgo importantes para el desarrollo y progresión de la misma, sus bases genéticas no están claras. Antes de 2008, los estudios sobre genes candidatos basados en conocimientos previos de la fisiopatología de la esteatosis hepática produjeron resultados contradictorios. En 2008, Romeo et al. publicaron el primer estudio de asociación amplia del genoma que presentaba indicios genéticos sólidos de la presencia de esteatosis hepática (PNPLA3, dominio de la fosfolipasa-patatina 3; rs738409). Desde entonces, se han publicado dos estudios adicionales de asociación amplia del genoma en los que se identificaron otras nueve variantes genéticas. Si bien estos resultados arrojan luz sobre la genética de la EHNA, la mayoría de las asociaciones no se han replicado en muestras independientes y, por tanto, la importancia de estos hallazgos continúa siendo incierta. Esta revisión pretende resumir los conocimientos actuales sobre la epidemiología genética de la EHNA y destaca las áreas de incertidumbre.
Nonalcoholic fatty liver disease (NAFLD), the most common chronic liver disease,1 represents a wide spectrum of disease characterized by the presence hepatic steatosis in the absence of significant alcohol consumption or other causes of liver disease.2,3 The pathogenic processes leading to steatosis, steatohepatitis (NASH) and fibrosis are multifactorial and involve both environmental and genetic factors.4,5 Obesity and type 2 diabetes/insulin resistance are the most common risk factors for the development and progression of NAFLD.6 Since both obesity and type 2 diabetes continue to rise, it is expected that NAFLD will reach epidemic proportions worldwide. The prevalence of ultrasound-defined NAFLD ranges from 14 to 45%.7 Extrapolating data from small studies and the current prevalence of obesity and type 2 diabetes, NAFLD was recently estimated to affect more than 30 million people in the U.S.8 In Spain, Caballeria et al. showed in a recent cross-sectional study a prevalence of 26% in individuals aged 15 and 85 years randomly selected from 25 primary healthcare centers,9 consistent with prior American estimates.
A genetic underpinning for NAFLD has been suggested by familial aggregation studies,10,11 heritability studies,12,13 candidate gene studies,4 genome-wide scans14–17 and expression studies.18–22 The presence of a genetic basis in NAFLD not only will shed light in the identification of individuals at risk to develop NAFLD and its progression, but also the dissection of NAFLD pathogenesis and the development of new therapies.
The ultimate goal of this review is to provide the big picture of the current understanding of genes associated with NAFLD so the reader is able to understand what the gaps are in the genetic epidemiology of NAFLD. This paper, however, will not assess other forms of fatty liver disease, expression studies or animal studies.
This work is structured in three parts: first, the author will provide some background concepts in the genetic epidemiology of NAFLD and the current understanding of NAFLD pathophysiology; second, the reviewer will highlight the most up-to-date findings in candidate gene studies and genome wide association studies (GWAS) in humans; the review will conclude with suggestions to conduct candidate gene studies. Compared to other reviews,23–25 the present work will not elaborate the mechanism for each specific gene but will provide references for the reader for a more careful evaluation.
Study designs and methodological problems in genetic epidemiology of NAFLDNAFLD is considered a complex disease because it does have a genetic component but with no simple Mendelian pattern of single-gene inheritance such as Wilson disease. In NAFLD, multiple genes, polygenes, environmental factors, age effects, and their interactions, may be involved.26
Genetic epidemiology studies can be divided into two broad categories: (a) according to the relatedness amongst participants (family-based versus non-related or population-based); and (b) according to the knowledge of the genetic marker(s) used (hypothesis-driven or candidate gene study versus hypothesis free or genome wide association study). A candidate gene is defined as gene whose product is considered to play a role in disease pathogenesis.27 It is important to bear in mind that all studies provide key information and are not mutually exclusive. For instance, family-based studies are typically the first step to determine if a condition is genetic in nature, the model of inheritance, and to understand the proportion of phenotypic variance due to shared genetic factors (heritability). Family-based studies may use linkage analysis to identify the location of a major gene or, association analyses such as the transmission-disequilibrium test, candidate gene approach and a genome-wide association analysis.
Population-based studies, on the other hand, is the mainstream of studies in NAFLD and, until recently, candidate gene studies were the most common design using the “two hit” hypothesis as a framework.5
Both candidate gene and GWAS rely on the statistical association of genetic markers and the disease (phenotype) of interest. The most commonly used genetic markers are the single nucleotide polymorphisms or SNPs, defined as the change in a single base pair of the DNA common in more than 1% of the population.23
The major problem in both candidate gene and GWAS is the presence of false negative and false positive associations. Lack of statistical power leads to false negatives; that is, true associations are not detected in the study. So the researcher needs to estimate the number of participants to be recruited to circumvent such error using power calculators. In GWAS, where there is no hypothesis behind the statistical analysis, the problem is the opposite and SNPs are spuriously associated due to chance with the disease, even after statistical correction for multiple comparisons. Consequently, most of the journals extremely recommend authors to replicate their findings in a different sample before accepting the new SNP as truly associated with NAFLD.
Steatosis: a protective mechanism surrounded by multiple insultsThe key pathological finding in fatty liver disease is the accumulation of triglycerides in the hepatocytes. This deposit is due to an imbalance between triglycerides acquisition and removal.28,29 Triglycerides are neutral lipids consisting of a glycerol backbone and three long-chain fatty acids. The major routes of free fatty acids (FFAs) are (a) dietary intake; (b) lipolysis from fat reservoirs, and (c) de novo lipogenesis. Animal studies and human inherited diseases have shown increased hepatic steatosis with increased triglyceride dietary intake, increased lipolysis from peripheral tissues, increased de novo lipogenesis, or prevention of triglyceride removal from the liver (i.e. decreased efflux or oxidation).28 The exact contribution of each pathway for the development of hepatic steatosis has been studied by Donnelly et al.30 who showed that 59% of hepatic fat derived from circulating FFAs (mainly lipolysis), 26% from de novo lipogenesis and 15% from diet.
Insulin resistance is the key etiopathogenic factor in the development of fatty liver. Whether insulin resistance is a cause of hepatic steatosis or vice versa is a matter of debate and has been reviewed recently.28 Current evidence suggest that insulin resistance comes first and leads to hepatic steatosis, as shown by animal models and human diseases where hepatic steatosis is not associated with insulin resistance and, conversely, human diseases with inherited insulin resistance leading to hepatic steatosis (e.g. AKT2 mutation).28 Opposite views, however, suggest that hepatic steatosis, possibly originated by impaired mitochondrial β-oxidation of fatty acids, leads to hepatic insulin resistance which, in turn, increases de novo fatty acid synthesis, and impairs glucogenogenesis, hepatic glucose uptake, and transport of lipoproteins perpetuating increase peripheral insulin resistance.7,29
In 1998, Day and James proposed a “two-hit model” for the development of steatohepatitis in which the first step was the accumulation of free fatty acids in the liver, and, from there, further insults induced inflammation, fibrosis and, eventually, cirrhosis.5 Nevertheless, Dr. Day's group updated their own model and suggested that “steatosis may be early adaptive response to hepatocyte stress through which potentially lipotoxic FFAs are partitioned into relatively stable intracellular triglyceride stores”.31 NAFLD is seen now “as a combination of effects of several fundamental biochemical and immunologic mechanisms of liver injury rather than adhering to a sequential ‘two-hit’ paradigm”31 and is being supported by other experts in the field.32 In Day et al.’s view, “insulin resistance promotes increased hepatic FFA flux (lipolysis, diet and lipogenesis) leading to hepatic steatosis, and is driven by (1) direct hepatocyte lipotoxicity, (2) hepatocellular oxidative stress secondary to free radicals produced during beta- and -FFA oxidation, (3) endotoxin/TLR4-induced inflammation, (4) cytokine release, and(5) endoplasmic reticulum (ER) stress”, leading, at the end, to inflammation, cellular damage, and progression to cirrhosis.
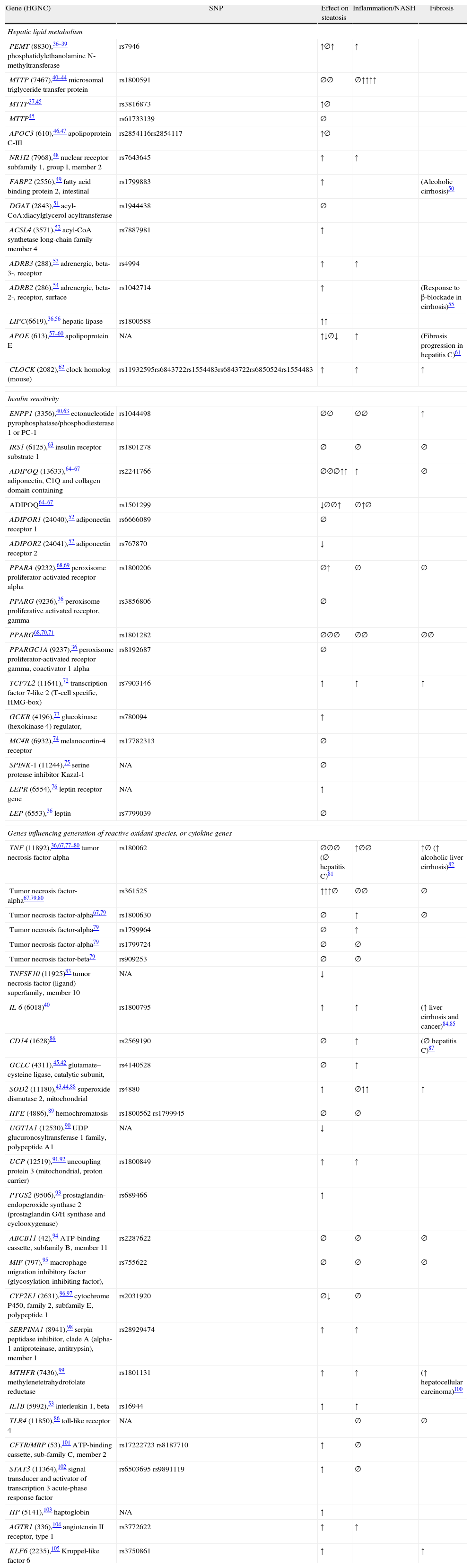
Genetic epidemiology of NAFLD: a systematic approachTo identify most of the available published literature in genetics of NAFLD, the author performed a systematic search using PUBMED by applying the following search engine: (polymorphism OR SNP) AND (“steatosis” OR “steatohepatitis” OR NAFLD OR NASH) without language restriction and until July 26th, 2011. The search yielded 224 references; of which 60 corresponded to candidate gene studies and are summarized in Table 1 and three genome wide association studies in Table 2. This search engine is limited by two major factors: on the one hand, it did not include EMBASE database (more sensitive for non-English papers and scientific meetings), and, on the other hand, there is still a risk by publication bias (negative studies are not published).
Candidate gene studies in nonalcoholic fatty liver disease (last updated 7/27/11).
| Gene (HGNC) | SNP | Effect on steatosis | Inflammation/NASH | Fibrosis |
| Hepatic lipid metabolism | ||||
| PEMT (8830),36–39 phosphatidylethanolamine N-methyltransferase | rs7946 | ↑∅↑ | ↑ | |
| MTTP (7467),40–44 microsomal triglyceride transfer protein | rs1800591 | ∅∅ | ∅↑↑↑↑ | |
| MTTP37,45 | rs3816873 | ↑∅ | ||
| MTTP45 | rs61733139 | ∅ | ||
| APOC3 (610),46,47 apolipoprotein C-III | rs2854116rs2854117 | ↑∅ | ||
| NR1I2 (7968),48 nuclear receptor subfamily 1, group I, member 2 | rs7643645 | ↑ | ↑ | |
| FABP2 (2556),49 fatty acid binding protein 2, intestinal | rs1799883 | ↑ | (Alcoholic cirrhosis)50 | |
| DGAT (2843),51 acyl-CoA:diacylglycerol acyltransferase | rs1944438 | ∅ | ||
| ACSL4 (3571),52 acyl-CoA synthetase long-chain family member 4 | rs7887981 | ↑ | ||
| ADRB3 (288),53 adrenergic, beta-3-, receptor | rs4994 | ↑ | ↑ | |
| ADRB2 (286),54 adrenergic, beta-2-, receptor, surface | rs1042714 | ↑ | (Response to β-blockade in cirrhosis)55 | |
| LIPC(6619),36,56 hepatic lipase | rs1800588 | ↑↑ | ||
| APOE (613),57–60 apolipoprotein E | N/A | ↑↓∅↓ | ↑ | (Fibrosis progression in hepatitis C)61 |
| CLOCK (2082),62 clock homolog (mouse) | rs11932595rs6843722rs1554483rs6843722rs6850524rs1554483 | ↑ | ↑ | ↑ |
| Insulin sensitivity | ||||
| ENPP1 (3356),40,63 ectonucleotide pyrophosphatase/phosphodiesterase 1 or PC-1 | rs1044498 | ∅∅ | ∅∅ | ↑ |
| IRS1 (6125),63 insulin receptor substrate 1 | rs1801278 | ∅ | ∅ | ∅ |
| ADIPOQ (13633),64–67 adiponectin, C1Q and collagen domain containing | rs2241766 | ∅∅∅↑↑ | ↑ | ∅ |
| ADIPOQ64–67 | rs1501299 | ↓∅∅↑ | ∅↑∅ | |
| ADIPOR1 (24040),52 adiponectin receptor 1 | rs6666089 | ∅ | ||
| ADIPOR2 (24041),52 adiponectin receptor 2 | rs767870 | ↓ | ||
| PPARA (9232),68,69 peroxisome proliferator-activated receptor alpha | rs1800206 | ∅↑ | ∅ | ∅ |
| PPARG (9236),36 peroxisome proliferative activated receptor, gamma | rs3856806 | ∅ | ||
| PPARG68,70,71 | rs1801282 | ∅∅∅ | ∅∅ | ∅∅ |
| PPARGC1A (9237),36 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha | rs8192687 | ∅ | ||
| TCF7L2 (11641),72 transcription factor 7-like 2 (T-cell specific, HMG-box) | rs7903146 | ↑ | ↑ | ↑ |
| GCKR (4196),73 glucokinase (hexokinase 4) regulator, | rs780094 | ↑ | ||
| MC4R (6932),74 melanocortin-4 receptor | rs17782313 | ∅ | ||
| SPINK-1 (11244),75 serine protease inhibitor Kazal-1 | N/A | ∅ | ||
| LEPR (6554),76 leptin receptor gene | N/A | ↑ | ||
| LEP (6553),36 leptin | rs7799039 | ∅ | ||
| Genes influencing generation of reactive oxidant species, or cytokine genes | ||||
| TNF (11892),36,67,77–80 tumor necrosis factor-alpha | rs180062 | ∅∅∅ (∅ hepatitis C)81 | ↑∅∅ | ↑∅ (↑ alcoholic liver cirrhosis)82 |
| Tumor necrosis factor-alpha67,79,80 | rs361525 | ↑↑↑∅ | ∅∅ | ∅ |
| Tumor necrosis factor-alpha67,79 | rs1800630 | ∅ | ↑ | ∅ |
| Tumor necrosis factor-alpha79 | rs1799964 | ∅ | ↑ | |
| Tumor necrosis factor-alpha79 | rs1799724 | ∅ | ∅ | |
| Tumor necrosis factor-beta79 | rs909253 | ∅ | ∅ | |
| TNFSF10 (11925)83 tumor necrosis factor (ligand) superfamily, member 10 | N/A | ↓ | ||
| IL-6 (6018)40 | rs1800795 | ↑ | ↑ | (↑ liver cirrhosis and cancer)84,85 |
| CD14 (1628)86 | rs2569190 | ∅ | ↑ | (∅ hepatitis C)87 |
| GCLC (4311),45,42 glutamate–cysteine ligase, catalytic subunit, | rs4140528 | ∅ | ↑ | |
| SOD2 (11180),43,44,88 superoxide dismutase 2, mitochondrial | rs4880 | ↑ | ∅↑↑ | ↑ |
| HFE (4886),89 hemochromatosis | rs1800562 rs1799945 | ∅ | ∅ | |
| UGT1A1 (12530),90 UDP glucuronosyltransferase 1 family, polypeptide A1 | N/A | ↓ | ||
| UCP (12519),91,92 uncoupling protein 3 (mitochondrial, proton carrier) | rs1800849 | ↑ | ↑ | |
| PTGS2 (9506),93 prostaglandin-endoperoxide synthase 2 (prostaglandin G/H synthase and cyclooxygenase) | rs689466 | ↑ | ||
| ABCB11 (42),94 ATP-binding cassette, subfamily B, member 11 | rs2287622 | ∅ | ∅ | ∅ |
| MIF (797),95 macrophage migration inhibitory factor (glycosylation-inhibiting factor), | rs755622 | ∅ | ∅ | ∅ |
| CYP2E1 (2631),96,97 cytochrome P450, family 2, subfamily E, polypeptide 1 | rs2031920 | ∅↓ | ∅ | |
| SERPINA1 (8941),98 serpin peptidase inhibitor, clade A (alpha-1 antiproteinase, antitrypsin), member 1 | rs28929474 | ↑ | ↑ | |
| MTHFR (7436),99 methylenetetrahydrofolate reductase | rs1801131 | ↑ | ↑ | (↑ hepatocellular carcinoma)100 |
| IL1B (5992),53 interleukin 1, beta | rs16944 | ↑ | ↑ | |
| TLR4 (11850),86 toll-like receptor 4 | N/A | ∅ | ∅ | |
| CFTR/MRP (53),101 ATP-binding cassette, sub-family C, member 2 | rs17222723 rs8187710 | ↑ | ∅ | |
| STAT3 (11364),102 signal transducer and activator of transcription 3 acute-phase response factor | rs6503695 rs9891119 | ↑ | ∅ | |
| HP (5141),103 haptoglobin | N/A | ↑ | ||
| AGTR1 (336),104 angiotensin II receptor, type 1 | rs3772622 | ↑ | ↑ | |
| KLF6 (2235),105 Kruppel-like factor 6 | rs3750861 | ↑ | ↑ | |
Abbreviations: N/A: not available; ↑: increase; ∅: no association; ↓: decrease (protective).
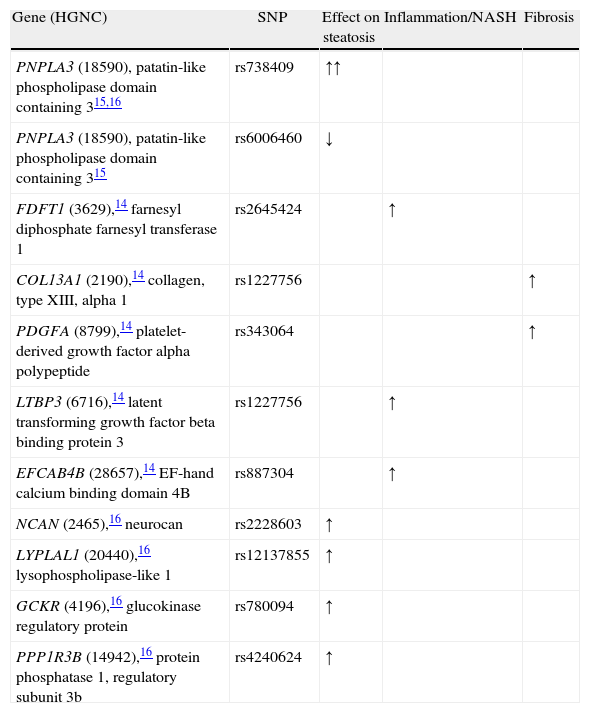
Genome-wide association studies in NAFLD (last updated 7/27/11).
| Gene (HGNC) | SNP | Effect on steatosis | Inflammation/NASH | Fibrosis |
| PNPLA3 (18590), patatin-like phospholipase domain containing 315,16 | rs738409 | ↑↑ | ||
| PNPLA3 (18590), patatin-like phospholipase domain containing 315 | rs6006460 | ↓ | ||
| FDFT1 (3629),14 farnesyl diphosphate farnesyl transferase 1 | rs2645424 | ↑ | ||
| COL13A1 (2190),14 collagen, type XIII, alpha 1 | rs1227756 | ↑ | ||
| PDGFA (8799),14 platelet-derived growth factor alpha polypeptide | rs343064 | ↑ | ||
| LTBP3 (6716),14 latent transforming growth factor beta binding protein 3 | rs1227756 | ↑ | ||
| EFCAB4B (28657),14 EF-hand calcium binding domain 4B | rs887304 | ↑ | ||
| NCAN (2465),16 neurocan | rs2228603 | ↑ | ||
| LYPLAL1 (20440),16 lysophospholipase-like 1 | rs12137855 | ↑ | ||
| GCKR (4196),16 glucokinase regulatory protein | rs780094 | ↑ | ||
| PPP1R3B (14942),16 protein phosphatase 1, regulatory subunit 3b | rs4240624 | ↑ |
Abbreviations: N/A: not available; ↑: increase; ∅: no association; ↓: decrease (protective).
Genes (or nearby genes) are named using the HUGO Gene Nomenclature Committee (HGNC) and reported identification.33
Candidate genes studies in human NAFLD: hepatic lipid metabolism, insulin resistance and oxidative stressNumerous candidate gene studies, applying this ‘multiple-hit’ hypothesis, have studied the effects of genes on NAFLD presence and progression. The reviewer has simplified the prior hypothesis into the following mechanisms (not mutually exclusive): genes involved in hepatic lipid metabolism (synthesis, storage, export, oxidation), genes implicated in insulin signaling (insulin resistance), and finally, genes involved in oxidative stress and inflammation (and therefore, most likely involved in progression to cirrhosis) (Table 1).
The reviewer included, when appropriate, other data from different phenotypes to illustrate the direction of association for a particular gene. The number of symbols represent how many studies have been done for a particular gene.
Genome wide association studies in NAFLDRomeo et al. were the first to apply the GWAS method using a phenotype based on magnetic resonance spectroscopy.15 They studied 9,299 nonsynonymous sequence variations and identified a missense mutation [Ile148→Met148 (I148M)] in patatin-like phospholipase domain-containing (PNPLA) 3 gene PNPLA3 (HCNG: 18590). PNPLA3, highly expressed in adipose tissue and liver, is regulated by insulin through a signaling cascade that includes LXR and SREBP-1c28 and, therefore, increased with feeding in animal studies.34 This mutation is, by far, the strongest genetic signal up to date and showed increased odds for fatty liver of 3.26 in the original report.15 In addition, the PNPLA3 gene could also be responsible for the difference in prevalence of fatty liver disease between ethnic groups. For instance, Mexican-Americans have more prevalence of the high risk allele, whereas African Americans, where fatty liver is known to be less frequent, had a protective variation for such.15 Future studies confirmed the association between this gene and the presence of fatty liver disease, including GWAS with liver enzymes17 and multiple case-control studies.35
Chalasani et al. described in 236 white female biopsy-proven NAFLD patients’ five new genetic variants associated with inflammation and fibrosis. They found that the NAFLD activity score (NAS), a pathological tool to measure changes in NAFLD during clinical trials, was associated with the gene FDFT1 (farnesyl diphosphate farnesyl transferase 1, HGNC: 3629); in addition, they found an association with lobular inflammation for the collagen gene COL13A1 (collagen, type XIII, alpha 1, HGNC: 2190), a SNP nearby the PDGFA gene (platelet-derived growth factor alpha polypeptide, HGNC: 8799); the LTBP3 (latent transforming growth factor beta binding protein 3, HGNC: 6716), and the EFCAB4B (EF-hand calcium binding domain 4B, HGNC: 28657).
The Genetics of Obesity-related Liver Disease (GOLD) Consortium is the last genome wide scan published up to date. We obtained the same association with PNPLA3 and four additional genetic variants including the PNPLA3 (HGNC: 18590), namely, NCAN (neurocan, HGNC: 2465), LYPLAL1 (lysophospholipase-like 1, HGNC: 20440); GCKR (glucokinase regulatory protein, HGNC: 4196); and the PPP1R3B (protein phosphatase 1, regulatory subunit 3b, HGNC: 14942). GCKR and PPP1R3B are key enzymes in de novo lipogenesis from glucose; LYPLAL1-related protein has been predicted to play a crucial part in consecutive steps in triglyceride breakdown. The role of NCAN, however, remains to be determined. Interestingly, we found that PNPLA3 and our other genes had a modest role for lipid metabolism suggesting that these genes, if they are involved in lipid metabolism, exert their effects within the liver through different mechanistic pathways than the observed ones by conventional laboratory lipid measurement.
There is no replication up to date of the aforementioned GWAS findings except for the PNPLA3 (rs738409). For this gene variant, a recent meta-analysis updated until January 2011 found an association with the presence of fat accumulation (GG homozygous showed 73% higher lipid fat content when compared with CC ones); 3.2-fold greater risk of higher necroinflammatory scores, 3.5-fold risk of NASH, and 3.2-fold greater risk of developing fibrosis when compared with CC homozygous.35
Future directions: candidate-genes are neededThe typical statement “more research is needed” is clearly shown by the examination of both tables. To better understand the genetic determinants of NAFLD, it is key to replicate prior studies. Whereas GWAS are expensive, require thousands of individuals and strong statistical and population genetics knowledge, the use of candidate gene studies in NAFLD is easy to implement and straightforward. The major caveat for these studies is the lack of power. For instance, assuming a genetic risk ratio of 1.5 for a given polymorphism (high for most of the replicated genetic studies), and a allele frequency about 20%, more than 400 patients and controls are required to give a study 90% power to detect a significant effect at the 5% level.27 Day27 and Bataller23 provide two outstanding reviews to guide the reader in the design of high quality candidate gene studies, step-by-step.
On the other hand, if the researcher is willing to invest his/her time to study new candidate genes, then Day proposes to find them by reviewing: “(1) Gene product considered to play a role in the disease; (2) gene is known to be mutated in a familial form of the disease; (3) gene knockout/overexpression in animal models influences disease development; (4) gene lies in a chromosomal region associated with disease in a linkage study; (5) gene expression is altered in microarray studies of tissue from patients with disease; (6) gene is identified in a phenotype-driven mouse mutagenesis study”27; and (7) gene identify in genome-wide association studies.
In conclusion, genes play a role in the development and progression of NAFLD; PNPLA3 is the strongest signal up to date but there are other numerous genes that have been described but not formally replicated. The future of genetic epidemiology will require replication and, ultimately, expression studies and animal models to know the molecular role of that particular genetic variant. The understanding of genetic determinants of NAFLD will help to identify individuals at risk and, potentially, new therapies to treat the most common chronic liver disease in the world.
Conflict of interestThe author declare no conflict of interest.
The author has been supported by the American Diabetes Association Mentor-Based Program (7-07-MN-08, PI: Dr. Frederick L. Brancati).

