






El síndrome hepatopulmonar está caracterizado por la existencia de enfermedad hepática, dilatación vascular pulmonar e hipoxemia arterial. Generalmente se asocia a una cirrosis hepática de cualquier origen aunque se ha descrito en otras enfermedades hepáticas, tanto agudas como crónicas, y no siempre asociada a la hipertensión portal. La ecocardiografía con contraste es el estándar de oro para el diagnóstico de las dilataciones vasculares pulmonares y fundamental por tanto para el diagnóstico del síndrome hepatopulmonar. Estas dilataciones reflejan cambios en la microvascularización pulmonar (vasodilatación, acúmulo intravascular de monocitos y angiogénesis) e inducen un desequilibrio en la relación ventilación/perfusión, o incluso verdaderos shunts, que finalmente desencadenan la hipoxemia. El síndrome hepatopulmonar empobrece el pronóstico y la calidad de vida de los pacientes y puede determinar la necesidad de un trasplante hepático que es el único tratamiento de eficacia demostrada. En el presente artículo se revisan los principales aspectos etiopatogénicos, fisiopatológicos, clínicos y terapéuticos de este síndrome.
Hepatopulmonary syndrome is characterized by the presence of liver disease, pulmonary vascular dilatations, and arterial hypoxemia. It is usually associated with cirrhosis of any origin, but has been described in other liver diseases, both acute and chronic, and not always associated with portal hypertension. The gold standard method to detect pulmonary vascular dilations is contrast enhancement echocardiography with saline and is essential for the diagnosis of hepatopulmonary syndrome. These dilatations reflect changes in the pulmonary microvasculature (vasodilatation, intravascular monocyte accumulation, and angiogenesis) and induce a ventilation/perfusion mismatch, or even true intrapulmonary shunts, which eventually trigger hypoxemia. This syndrome worsens patients’ prognosis and impairs their quality of life and may lead to the need for liver transplantation, which is the only effective and definitive treatment. In this article, we review the etiological, pathophysiological, clinical and therapeutic features of this syndrome.
El síndrome hepatopulmonar (SHP) se describió por primera vez en 1977 y está caracterizado por una tríada que comprende la existencia de enfermedad hepática, dilatación vascular pulmonar e hipoxemia arterial1–5. Se asocia con mayor frecuencia a la existencia de cirrosis hepática de cualquier origen aunque se ha descrito en otras enfermedades hepáticas, tanto agudas como crónicas, y no siempre asociada a la presencia de hipertensión portal2–5. Su presencia empobrece el pronóstico y la calidad de vida de los pacientes y puede determinar la necesidad de un trasplante hepático (TH) que es el único tratamiento de eficacia demostrada para estos pacientes2–7. En los siguientes apartados se realiza una revisión de los principales aspectos etiopatogénicos, fisiopatológicos, clínicos y terapéuticos de este síndrome.
DefiniciónEl SHP se caracteriza por un defecto en la oxigenación arterial inducida por una dilatación vascular pulmonar en el contexto de una enfermedad hepática avanzada y/o hipertensión portal2–7. Así pues, este síndrome clínico tiene 3 componentes: defecto en la oxigenación, enfermedad hepática y dilatación vascular pulmonar.
La definición de hipoxemia asociada al SHP se basa en la medición de la presión parcial arterial de oxígeno (PaO2) del paciente en una posición estandarizada, preferiblemente en decúbito y sentado. Sin embargo, la determinación del gradiente alvéolo-arterial de oxígenoc (PA-aO2) es un método más sensible para detectar defectos de oxigenación ya que puede incrementarse anormalmente antes de que la PaO2 caiga por debajo de la normalidad (su medición compensa los niveles reducidos de dióxido de carbono, la hiperventilación y la alcalosis respiratoria observada habitualmente en los pacientes con enfermedad hepática avanzada). Se ha establecido como consenso para el diagnóstico de SHP un límite mayor o igual a 15mmHg en menores de 65 años y mayor o igual a 20 en mayores de 65 años2,5,6. La pulsioximetría para la valoración de la saturación arterial de oxígeno (SatO2) es útil en el seguimiento de los pacientes con SHP moderado o severo e incluso puede aportar información útil ya que una SatO2 ≥96% excluye una PaO2<70mmHg8, pero no es lo suficientemente exacta para reemplazar la información que proporciona la gasometría arterial. Por otra parte, hay que recordar que la hipoxemia arterial es común en el contexto de la enfermedad hepática, pudiendo ser de causa multifactorial (ascitis, hidrotórax hepático, enfermedad pulmonar obstructiva crónica en pacientes con alcoholismo crónico, etc.)4,9. Es característica del SHP una respuesta completa de la PaO2 hasta 300mmHg a un flujo de O2 del 100%10, excepto en los casos en los que existen comunicaciones arteriovenosas (SHP tipo II; ver apartado Diagnóstico), y en algunos casos graves o muy graves debido a los cambios en la pared del vaso que alteran la difusión11,12. Ya que, como se acaba de comentar, los pacientes con enfermedad hepática avanzada hiperventilan, es frecuente observar hipocapnia (PaCO2<35mmHg) y alcalosis respiratoria. Por el contrario, no se observa nunca retención de dióxido de carbono.
El componente vascular incluye la dilatación de capilares pulmonares difusa o localizada y, menos frecuentemente, la presencia de comunicaciones arteriovenosas pleurales y pulmonares e incluso verdaderas anastomosis venosas portopulmonares4,13. Sin embargo, la dilatación de vasos capilares en regiones alveolares es un prerrequisito y el principal rasgo distintivo fisiopatológico de la desoxigenación arterial en el SHP, tanto en humanos como en modelos experimentales. Como se describe más adelante, la realización de una ecocardiografía con realce de contraste (ECC) con salino (agitado para producir microburbujas de>10μm de diámetro) constituye el método más práctico para detectar una dilatación vascular pulmonar y es considerado como el principal método de cribado y de diagnóstico2,14. La gammagrafía pulmonar con macroagregados de albúmina marcados con 99mTc (γMA) es un método alternativo, más específico pero menos sensible15.
El tercer componente de la tríada que define al SHP es la existencia de una enfermedad hepática. El SHP aparece en el seno de una enfermedad hepática, tanto aguda como crónica, principalmente en la cirrosis hepática, la enfermedad hepática que más frecuentemente se asocia a este síndrome2–7. El hecho de que el SHP pueda ocurrir en el seno de enfermedades hepáticas agudas y en hepatitis crónicas no cirróticas, demuestra que la hipertensión portal puede no estar presente en todos los casos2,9,10. Puede ocurrir con cualquier grado de enfermedad hepática desde una enfermedad hepática crónica compensada sin cirrosis a una hipertensión portal no cirrótica o cirrosis, habiéndose descrito también en el fallo hepático agudo16–18. Sin embargo, aunque no se requiere la existencia de hipertensión portal (HTP) para que el SHP se manifieste, la mayoría de los casos de SHP se asocian con signos clínicos de HTP cirrótica o no cirrótica (ej. varices esofágicas, esplenomegalia o ascitis)2–7. La etiología de la enfermedad hepática no parece tener importancia como causante de la vasodilatación pulmonar, y se ve asociada a trastornos de naturaleza diferente: además de la cirrosis hepática se ha visto asociada a hepatitis granulomatosa, síndrome de Budd-Chiari, hepatitis fulminante y hepatitis isquémica. Por otra parte, no parece existir una relación clara entre la presencia o gravedad del SHP y la gravedad de la enfermedad hepática valorada por la clasificación de Child-Turcotte-Pugh o por el índice MELD (Model of End-Stage Liver Disease). Aunque se ha comunicado sobre todo en pacientes con enfermedad hepática gravemente descompensada, principalmente en estadio C de Child (realmente el grupo donde se realiza un cribado más intenso), también se ha observado en pacientes con Child A y B14,19.
EpidemiologíaEl SHP se observa principalmente en pacientes de mediana edad (generalmente diagnosticado en la sexta década de la vida) sin diferencia entre sexos aunque también puede verse en niños. La prevalencia del SHP varía entre las series, según los criterios y los métodos diagnósticos utilizados y la población estudiada, entre el 4 y el 44%2,20–22. En pacientes con cirrosis, la prevalencia media de SHP es del 15%, dependiendo del valor de corte para la PaO2 utilizado para definir esta entidad y/o del medio de cálculo del PA-aO2. En enfermedades hepáticas raras se desconoce la prevalencia pero se estima que debe de ser muy baja a tenor de la incidencia de estas enfermedades. En hepatitis crónicas virales, con o sin cirrosis, la prevalencia se ha situado alrededor del 10%. En pacientes candidatos a TH, la prevalencia es elevada (18-30%)8,22,23 e incluso mayor en pacientes con el síndrome de Budd-Chiari21. El SHP suele ser un proceso infradiagnosticado, debido principalmente a la coexistencia de enfermedad hepática (por lo general, cirrosis) que conlleva una alteración fisiopatológica pulmonar capaz de justificar los trastornos de oxigenación y la sensación disneica que presentan algunos de estos pacientes, así como al desconocimiento general de una entidad poco estudiada, con fisiopatología compleja y criterios diagnósticos dispersos hasta la actualidad, incluso para ambientes médicos especializados. Para reducir la variabilidad y establecer un consenso en cuanto al diagnóstico, un comité de expertos, la Task Force on Pulmonary-Hepatic Vascular Disorders (TFPHVD) de la European Respiratory Society ha propuesto un algoritmo para el diagnóstico y el manejo del SHP que es la base sobre la que distintos grupos trabajan con la introducción de alguna variante tal y como se comenta más adelante2,5,6.
Fisiopatología y etiopatogeniaLa causa de la hipoxemia en el SHP es la marcada dilatación de los vasos precapilares y poscapilares junto con un incremento en el número de vasos dilatados. Además, se pueden observar algunas comunicaciones arteriovenosas pulmonares y pleurales (shunts) y anastomosis portopulmonares2,13. Todo ello, permite que la sangre venosa hipooxigenada pase rápidamente a las venas pulmonares, con la consiguiente disminución de la oxigenación de la sangre arterial13. Los tres mecanismos intrapulmonares causantes de la hipoxemia en el SHP son: a) el desequilibrio de la relación ventilación/perfusión causado por la existencia de alvéolos hiperperfundidos; b) el cortocircuito o shunt intrapulmonar causado por áreas de desequilibrio de ventilación/perfusión extrema, o incluso la existencia de verdaderas fístulas arteriovenosas intrapulmonares documentadas en escasos estudios en autopsias, y c) el fracaso de la disminución de la difusión de oxígeno al tener que recorrer las moléculas de oxígeno un espacio ampliado por la vasodilatación pulmonar (defecto de la difusión-perfusión)4. Además, un estado de circulación hiperdinámica puede condicionar una circulación pulmonar más rápida, que puede dificultar el intercambio gaseoso. Los estadios iniciales y leves del SHP, caracterizados solo por un PA-aO2 aumentado o una PaO2 entre 60 y 80mmHg mientras el paciente respira aire ambiente, están causados principalmente por un desequilibrio en la relación ventilación-perfusión con o sin un shunt modesto (< 10%). Este parece ser el principal mecanismo de hipoxemia en el SHP lo cual está apoyado por la mejoría en la PaO2 en la mayoría de los pacientes con oxígeno al 100%. En los estadios más avanzados del SHP grave (ej. PaO2 <60mmHg) pueden intervenir todos los mecanismos intrapulmonares determinantes de un intercambio gaseoso anómalo.
Aunque muchos investigadores consideran que la causa íntima del SHP es la HTP, no se ha demostrado en el modelo de experimentación animal, ya que la oclusión parcial de la porta en ratas, que produce HTP y circulación hiperdinámica pero no daño hepático, no conduce a SHP24. Como ya se ha comentado, la presencia de HTP no se requiere para que se manifieste el SHP en el humano. Por otra parte, la correlación entre el grado de disfunción hepática y de HTP, y la prevalencia y gravedad del SHP es controvertida2. Aunque hay discordancias en la literatura médica, varios trabajos señalan que la gravedad de la enfermedad hepática guarda relación con la prevalencia de SHP25–27.
Las principales características fisiopatológicas del SHP se conocen a través del modelo experimental de cirrosis biliar inducida en ratas mediante la ligadura del conducto biliar común (LCBC) en ratas, principal y más utilizado modelo animal del SHP. Este modelo animal produce una cirrosis biliar y reproduce los mecanismos fisiopatológicos del SHP. Las principales alteraciones objetivadas mediante este modelo residen en la microvascularización pulmonar, incluyendo la vasodilatación, el acúmulo intravascular de monocitos y la angiogénesis22,28–37.
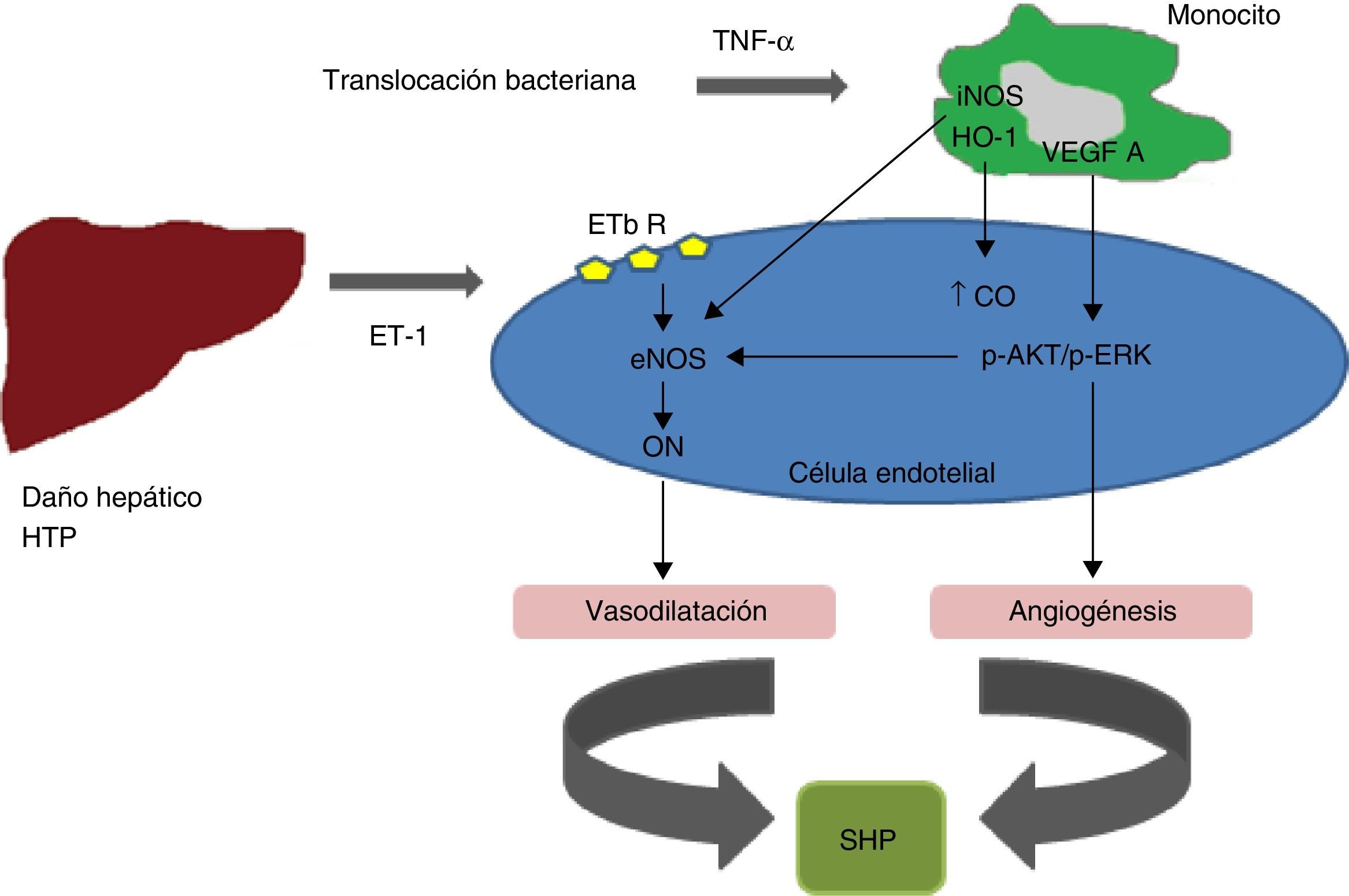
La visión fisiopatológica actual del SHP implica varios mediadores en la génesis de la vasodilatación pulmonar. En pacientes cirróticos con SHP se ha demostrado un incremento de la producción de óxido nítrico (ON) en los pulmones, medido en el aire espirado, que se corrige tras el TH36. Sin embargo, el mecanismo del incremento de la producción endógena de ON y su relación con la presencia de HTP, la circulación hiperdinámica y el grado de daño hepático es aún desconocido. Por otra parte, el uso de inhibidores del óxido nítrico en el tratamiento del SHP ha producido resultados discrepantes4. En el modelo de LCBC se ha demostrado un aumento en la expresión de la sintetasa del óxido nítrico, tanto de la forma inducible (iNOS, en macrófagos) como de la constitutiva endotelial (eNOS) como origen del incremento en la producción del ON pulmonar28. También se ha demostrado que ligeros aumentos de la producción hepática y liberación de pequeñas cantidades de endotelina-1 es uno de los mecanismos que desencadenan un incremento en la producción de eNOS y vasodilatación tras la LCBC34. Esto se asocia a un incremento selectivo de la expresión de los receptores B de la endotelina-1 (ETB) a nivel vascular pulmonar en la cirrosis y HTP, que finalmente contribuyen a la producción aumentada de ON29 (fig. 1).
, que resultan en la producción o liberación de mediadores en la circulación venosa, a cuyo nivel influyen en la microcirculación pulmonar. Una expresión aumentada de los receptores pulmonares endoteliales ETB y un aumento en la producción y liberación hepática de ET-1 contribuyen a un incremento en la expresión de eNOS y en la producción de óxido nítrico (ON) en el endotelio microvascular durante la iniciación del SHP. La acumulación intravascular de células macrófago-like mediada por el factor de necrosis tumoral (TNFα) también ocurre tras la ligadura crónica del conducto biliar común (LCBC). Se produce un incremento de la expresión de la Haem oxigenasa (HO)-1 y la sintetasa inducible del ON (iNOS) en estas células lo que también contribuye a la progresión del SHP. eNOS: óxido nítrico sintetasa endotelial; ET-1: endotelina 1; ETbR: receptor b de la endotelina; HO-1: hemo-oxigenasa 1; iNOS: óxido nítrico sintetasa inducible; OC: monóxido de carbono; SHP: síndrome hepatopulmonar; TNFα: factor de necrosis tumoral alfa; VEGF-A: factor de crecimiento endotelial vascular A.")
Modelo conceptual de la patogénesis del SHP. Durante el desarrollo del SHP, se producen una serie de alteraciones, directa e indirectamente relacionadas con el daño hepático y la hipertensión portal (HTP), que resultan en la producción o liberación de mediadores en la circulación venosa, a cuyo nivel influyen en la microcirculación pulmonar. Una expresión aumentada de los receptores pulmonares endoteliales ETB y un aumento en la producción y liberación hepática de ET-1 contribuyen a un incremento en la expresión de eNOS y en la producción de óxido nítrico (ON) en el endotelio microvascular durante la iniciación del SHP. La acumulación intravascular de células macrófago-like mediada por el factor de necrosis tumoral (TNFα) también ocurre tras la ligadura crónica del conducto biliar común (LCBC). Se produce un incremento de la expresión de la Haem oxigenasa (HO)-1 y la sintetasa inducible del ON (iNOS) en estas células lo que también contribuye a la progresión del SHP.
eNOS: óxido nítrico sintetasa endotelial; ET-1: endotelina 1; ETbR: receptor b de la endotelina; HO-1: hemo-oxigenasa 1; iNOS: óxido nítrico sintetasa inducible; OC: monóxido de carbono; SHP: síndrome hepatopulmonar; TNFα: factor de necrosis tumoral alfa; VEGF-A: factor de crecimiento endotelial vascular A.
Por otra parte, se han observado aumentos de la eNOS y la iNOS así como un aumento de los macrófagos en el lecho intravascular pulmonar en respuesta a incrementos del factor de necrosis tumoral alfa (TNF-α), resultado de la frecuente translocación bacteriana30,31. El papel del TNF-α se ha sugerido en modelos experimentales en los cuales se evitó la aparición del SHP con pentoxifilina, un inhibidor de la producción del TNF-α37. Por otra parte, la administración de antibióticos como el norfloxacino como tratamiento tras la LCBC reduce la acumulación de macrófagos y normaliza la iNOS pero no la eNOS, lo que apoya el papel de la translocación bacteriana en la acumulación de macrófagos a nivel pulmonar y su contribución a la dilatación venosa intrapulmonar30. La endotelina-1, además de incrementar la activación de la eNOS, contribuye a la acumulación de monocitos a través de la sobreexpresión de endotelina B en el endotelio vascular pulmonar. La translocación bacteriana y la endotoxinemia también contribuyen a la acumulación de macrófagos en la microvascularización pulmonar. La activación endotelial de la citokina fractalkina (CX3CL1) en el pulmón puede ser una vía común para la adherencia de los monocitos en la microcirculación pulmonar30–32. Los monocitos expresan iNOS y también producen heme oxigenasa-1 lo que conduce a un incremento en la producción de CO y, por lo tanto, a un aumento de la vasodilatación35.
Más recientemente, se ha reconocido que la angiogénesis pulmonar contribuye a jugar un papel importante en las alteraciones vasculares del SHP experimental32,33. Los monocitos de la microvascularización pulmonar producen factores de crecimiento como el VEGF-A (factor A de crecimiento vascular endotelial) que contribuye a la angiogénesis mediante la activación de vías de señalización que incluyen Akt y ERK en las células endoteliales6. La señalización mediada por CX3CL1/CX3CR1 contribuye al acúmulo de monocitos y la angiogénesis.
Finalmente, se ha observado que algunos polimorfismos de nucleótido único (SNP) en genes involucrados en la regulación de la angiogénesis están asociados al riesgo de SHP en cirróticos38. Concretamente, Roberts et al. llevaron a cabo un estudio de casos y controles sobre 59 casos con SHP diagnosticado con los criterios ya conocidos y 126 controles en los que se realizó un genotipado de 1086 SNP en 94 genes candidatos involucrados en la regulación de la angiogénesis. Encontraron una asociación de estos polimorfismos con el riesgo de desarrollar SHP. En resumen, encontraron 42 SNP en 21 genes que estaban significativamente asociados al SHP tras los ajustes por raza y tabaco. Ocho genes tuvieron al menos 2 SNP asociados con la enfermedad: CAV3, ENG, NOX4, ESR2, VWF, RUNX1, COL18A1 y TIE1. Por ejemplo, rs4837192 en CAV3 mostró una odds ratio de 2,75 (IC95% 1,65-4,6) y el rs4837192 en ENG mostró una odds ratio de 0,35 (IC95% 0,14-0,89). Además, la variación en CAV3 y RUNX1 se asoció con el SHP.
ClínicaLas manifestaciones clínicas del SHP dependen de la intensidad del trastorno gaseoso. En estadios leves los pacientes no suelen presentar disnea y predominan la astenia y otras manifestaciones de la enfermedad hepática. La disnea durante el ejercicio físico, en reposo o en ambas situaciones, constituye el síntoma predominante, generalmente después de años de enfermedad hepática y en estadios avanzados del SHP2–7. No se trata de un síntoma específico y puede encontrarse con frecuencia en estos pacientes debido a distintas complicaciones hepáticas como la anemia, la ascitis y retención líquida o la debilidad muscular. Aunque no hay síntomas o signos patognomónicos del SHP, la presencia de arañas vasculares, acropaquias, cianosis e hipoxemia severa (< 60mmHg) sugieren con fuerza un SHP39. También es característica la presencia de ortodeoxia (caída de un 5% o más de 4mmHg en la PaO2 cuando el paciente cambia de la posición decúbito supina al ortostatismo) lo cual se traduce en una mayor disnea (platipnea) relacionada con un desequilibrio de la ventilación-perfusión. Aunque no es específico del SHP, representa un mayor grado de vasodilatación pulmonar. Diversos estudios sitúan la prevalencia de este síntoma en pacientes con SHP en un 25%40; sin embargo, en un reciente estudio23 se observa que la presencia de la ortodeoxia es similar en los pacientes con y sin SHP (30,4 vs. 32,9%), sin diferencias estadísticamente significativas sobre una muestra de 199 pacientes. En cualquier caso, estas manifestaciones clínicas clásicas del SHP no son patognomónicas y guardan una pobre correlación con la severidad de la enfermedad hepática de manera que, aunque se han descrito principalmente en pacientes con cirrosis Child C, también se han observado en pacientes con cirrosis estadio Child A o B19,41.
DiagnósticoPruebas diagnósticasLas herramientas disponibles para el diagnóstico del componente vascular del SHP son principalmente la ECC y la γMA. La gasometría arterial nos permitirá definir el componente de hipoxemia tal y como se ha definido con anterioridad. Además, nos permitirá evaluar la gravedad del SHP según el grado de hipoxemia, siempre con un PA-aO2 ≥ 15mmHg (≥ 20mmHg en mayores de 65 años), (tabla 1). Anteriormente se ha descrito el papel de la pulsioximetría en el diagnóstico y cribado del SHP.
Niveles de gravedad del síndrome hepatopulmonar (SHP) según el grado de hipoxemia. En todos los casos el gradiente alvélolo-arterial de oxígeno (PA-aO2), parámetro más sensible para detectar la hipoxemia, para el diagnóstico de SHP es ≥ 15mmHg (para adultos menores de 65 años) y ≥ 20mmHg en adultos mayores de 65 años (ver texto)
| Nivel de gravedad | Hipoxemia |
|---|---|
| Leve | Presión parcial de oxígeno ≥ 80mmHg |
| Moderado | Presión parcial de oxígeno ≥ 60mmHg y<80mmHg |
| Grave | Presión parcial de oxígeno ≥ 50mmHg y<60mmHg |
| Muy grave | Presión parcial de oxígeno<50mmHg |
La realización de una ECC con salino constituye el método más práctico para detectar una dilatación vascular pulmonar. Este método cualitativo es más sensible y menos invasivo, aunque menos específico para el diagnóstico del SHP que la γMA2–7. Actualmente se considera la ECC el estándar de oro para el diagnóstico de las dilataciones vasculares pulmonares en el SHP y es el método de cribado cualitativo4,14. Tras la administración de salino agitado a través de una vena periférica del brazo, la existencia de una opacidad de la aurícula izquierda dentro de 3 a 6 ciclos cardiacos después de la opacidad de la aurícula derecha indica el paso de microburbujas a través de un árbol vascular anormalmente dilatado; las microburbujas no pasan a través de los capilares normales cuyo diámetro normal varía en un rango entre 8-15μm por lo que solo son visibles en las cavidades derechas. En el SHP las burbujas pasan a través de los capilares pulmonares dilatados, alcanzan la circulación general y son también visibles en las cavidades izquierdas del corazón (cortocircuito derecha-izquierda). Este método presenta algunas interrogantes como son el significado de la aparición del eco con contraste en la aurícula izquierda más allá del sexto latido, y por otro lado, el que hasta un 11-47% de los pacientes con cirrosis (con o sin SHP asociado) pueden presentar una ECC positiva, sin que tengan un PA-aO2 elevado ni hipoxemia2,14,39. Estos pacientes pueden representar una forma larvada o silente de SHP cuya evolución es desconocida4.
La γMA se basa en que las moléculas de albúmina marcadas 99mTc, al tener un tamaño 4 veces mayor que el capilar pulmonar en condiciones normales quedan atrapadas en el lecho capilar pulmonar. La relación de cuentas radiactivas entre el pulmón y un órgano de circulación estable como el cerebro permite diagnosticar la existencia de vasodilatación pulmonar: Una captación cerebral superior al 6% es indicativa de una dilatación vascular pulmonar y se observa en todos los casos de SHP con PaO2<60mmHg; este límite presenta una sensibilidad del 84% y una especificidad del 100% en el diagnóstico del SHP15. Por otra parte, esta técnica permite cuantificar el grado de cortocircuito intrapulmonar y no se ve afectado por otras enfermedades respiratorias asociadas. Su principal aplicación es la de cuantificar el grado de shunt y determinar que la hipoxemia se debe principalmente al SHP cuando coexisten una dilatación vascular pulmonar y otras enfermedades respiratorias que contribuyen a la alteración de la oxigenación arterial (PaO2≤ 60mmHg) si la captación cerebral es mayor al 6%11; por último, de cara al pronóstico, una captación cerebral ≥ 20% en pacientes con SHP grave (PaO2 ≤ 50mmHg) predice una elevada mortalidad postrasplante12. La mayor desventaja de la γMA es su incapacidad para diferenciar entre dilataciones venosas intrapulmonares y verdaderas comunicaciones intracardiacas. Por el contrario, la ECC permite diferenciar si el cortocircuito es intrapulmonar o intracardiaco mediante la observación del momento del paso de microburbujas: si es precoz (< 3 ciclos cardiacos) indicaría cortocircuito intracardiaco, mientras que si es tardío (entre 3 y 6 ciclos cardiacos tras la inyección), es diagnóstico de cortocircuito intrapulmonar. Otro método que permite esta diferenciación es la ecocardiografía transesofágica realzada con contraste que identifica directamente el septo interauricular, la posible existencia de un shunt derecha-izquierda, y muestra directamente el paso de microburbujas hacia la aurícula izquierda a través de un defecto del septo42. Este último método puede aumentar la sensibilidad en casos de duda con el ECC.
No obstante, ninguno de los métodos es válido para diferenciar entre comunicaciones arteriovenosas discretas (SHP tipo II) y dilataciones precapilares y capilares difusas (SHP tipo I); las primeras son anatómicamente estables y pueden no regresar tras el trasplante causando hipoxemia persistente postrasplante5. Esta distinción se puede hacer mediante una angiografía pulmonar. La angiografía, no obstante, tiene un papel restringido y no se considera en la actualidad una herramienta necesaria para el diagnóstico. En el SHP, la angiografía solo debería realizarse cuando la hipoxemia es severa (ej. PaO2<60mmHg), responde mal a la administración de oxígeno al 100%, y cuando hay una sospecha alta mediante TAC torácica de la existencia de comunicaciones arteriovenosas directas susceptibles de embolización5. La TAC de alta resolución torácica puede proporcionar información sobre la forma y distribución de los vasos pulmonares pero su exactitud diagnóstica no está suficientemente establecida.
En cuanto a las pruebas de función respiratoria (PFR), tanto los volúmenes estáticos pulmonares como los resultados de la espirometría forzada están de forma característica dentro de los valores normales en el SHP, siempre y cuando no haya otra comorbilidad pulmonar2–7. La capacidad de difusión del monóxido de carbono (DLCO) es el único test de función pulmonar rutinario anormal de forma consistente en el SHP pero no es específico y no se recomienda como herramienta diagnóstica ni de cribado39. No se precisa la biopsia pulmonar para el diagnóstico ya que las dilataciones vasculares intrapulmonares no se detectan de forma fiable en muestras tisulares.
Recientemente se ha propuesto la medición de los niveles séricos en sangre periférica del antígeno del factor de Von Willebrand (Ag-FvW), un marcador de disfunción endotelial, como método de cribado del SHP43. Estos investigadores observaron en una cohorte de 125 pacientes con cirrosis compensada que los niveles de Ag-FvW aparecían elevados en pacientes con SHP frente a pacientes sin SHP, y esto se correlacionaba con el defecto de la oxigenación. En dicho estudio, se observaba también que el área bajo la curva del Ag-FvW era significativamente más predictivo para estadios leves moderados del SHP en comparación con la SatO2. Sin duda se trata de una herramienta cuya utilidad debe ser ratificada en futuros estudios.
Estrategias de diagnósticoSegún la Task Force on Pulmonary-Hepatic Vascular Disorders anteriormente mencionada, todo paciente con enfermedad hepática, con un PA-aO2 incrementado para su edad, con o sin hipoxemia y con una ECC positiva debe considerarse afectado de SHP2,5. La existencia de otra enfermedad intrínseca pulmonar no excluye el diagnóstico aunque será necesario evaluar la contribución del SHP a la hipoxemia del paciente.
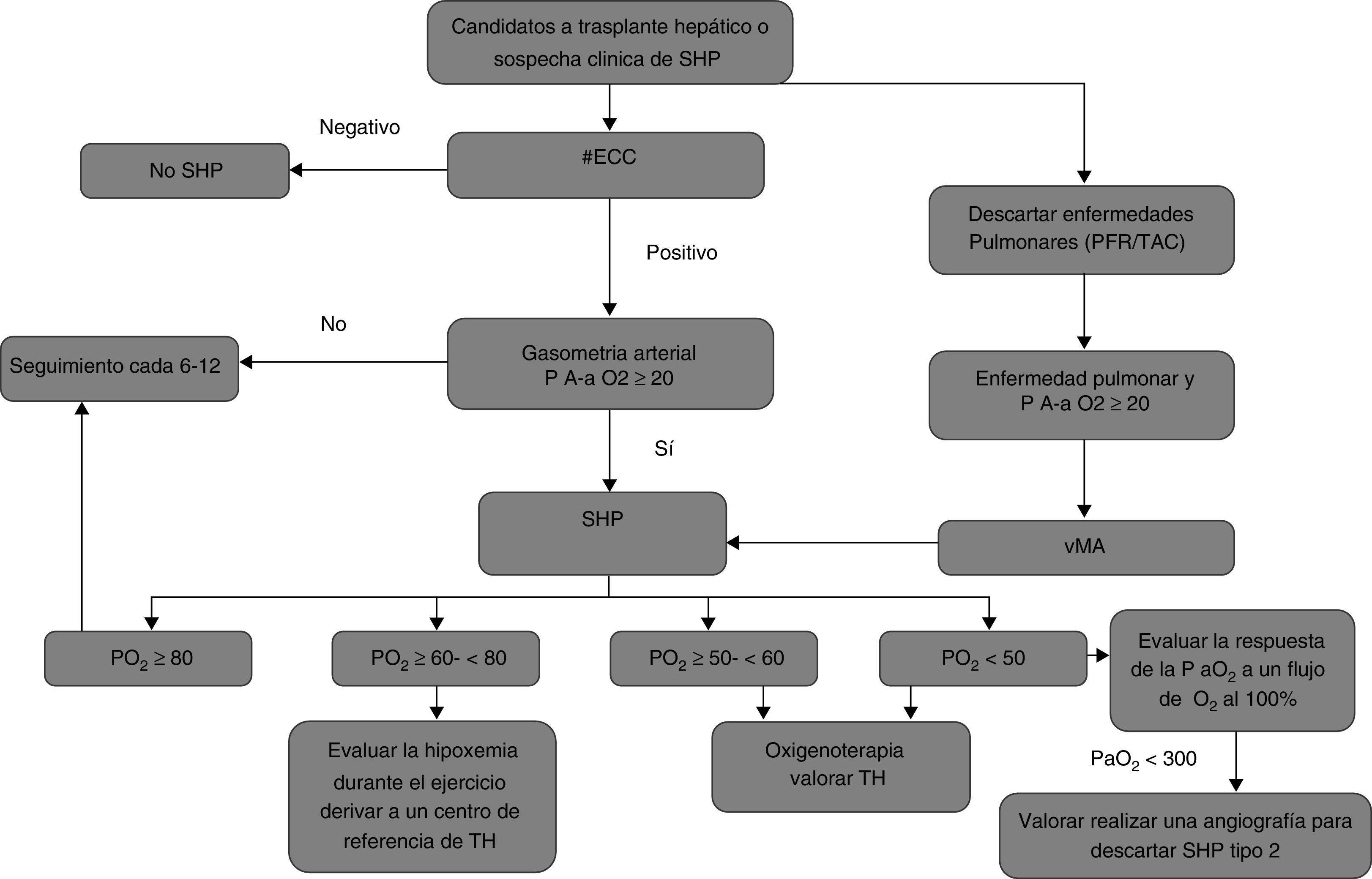
Este organismo recomienda, particularmente para todos los pacientes candidatos a TH y para los pacientes con enfermedad hepática con disnea o síntomas compatibles, llevar a cabo un algoritmo diagnóstico para detectar y tratar el SHP. En las guías del año 20042 proponen la realización de una gasometría arterial incluyendo el cálculo del PA-aO2. Si se confirma un PA-aO2 ≥ 15mmHg, con o sin hipoxemia, se debe realizar una ECC y una batería completa de PFR incluyendo una espirometría forzada con respuesta broncodilatadora, cálculo de volúmenes estáticos y DLCO. Una ECC negativa excluye el diagnóstico de SHP mientras que una ECC positiva establece el diagnóstico del SHP independientemente de la presencia o ausencia de una enfermedad cardiopulmonar intrínseca. En una revisión posterior publicada en el año 20125 se propone comenzar directamente con una ECC en la población referida siguiendo el algoritmo diagnóstico expuesto en la figura 2. Este algoritmo está basado en los protocolos de TH de la Clínica Mayo y las recomendaciones de la Sociedad Europea de Neumología5. Una ECC negativa descarta el diagnóstico mientras una ECC positiva obliga a la realización de una gasometría arterial para determinar el PA-aO2. Si este es menor a 20mmHg, se debe realizar una gasometría anual para hacer un seguimiento despistaje del SHP en la evolución. En el caso de un gradiente ≥ 20mmHg, se confirma el SHP y se procede a caracterizarlo en cuanto a la gravedad según el grado de hipoxemia. Si existe clínica respiratoria y/o alteraciones en la oxigenación gasométrica (hipoxemia y/o alteración en el gradiente A-a), se debe además realizar un estudio apropiado de despistaje de una enfermedad respiratoria mediante la realización de unas PFR, radiografía de tórax y, preferentemente de una TAC torácica de alta resolución independientemente. De forma complementaria se puede calcular el índice de perfusión mediante una γMA para estimar el grado de vasodilatación pulmonar y el pronóstico tras el trasplante; además, como se ha comentado, esta prueba es útil para confirmar que la causa de la hipoxemia es el SHP cuando también existe otra enfermedad pulmonar. Finalmente, se establece un algoritmo de manejo terapéutico según la gravedad del SHP (tabla 1 y fig. 2).

Algoritmo para el cribado y manejo terapéutico del síndrome hepatopulmonar propuesto por Porres-Aguilar et al.
ECC: ecocardiografíacon realce de contraste; γMA: gammagrafía con macroagregados de albúmina marcados con 99mTc; P A-aO2: gradiente alvéolo-arterial de oxígeno; Pa O2: presión arterial de oxígeno; PFR: pruebas de función respiratoria; TAC: tomografía axial computarizada; TH: trasplante hepático; #: si existe duda sobre un defecto del septo auricular, se recomienda un ecocardio transesofágico.
Fuente: Porres-Aguilar et al.5.
En los últimos años, la pulsioximetría para la medición de la SatO2 ha emergido como un método asequible, de bajo coste y fiable para el cribado del SHP en pacientes cirróticos ya que permite diagnosticar un grado moderado del SHP con una buena sensibilidad y especificidad (si la Sat O2 es ≤ 94% se pueden detectar pacientes con SHP con una PaO2 <60mmHg con una sensibilidad del 100% y una especificidad del 93%)8; se trataría de un método coste-efectivo para el cribado de SHP en pacientes en lista de espera para TH44. En este sentido, el grupo de Machicao et al., propone un algoritmo diagnóstico del SHP en candidatos a TH basado en la pulsioximetría con posterior realización de un ECC6. Sin embargo, la mayor limitación de esta técnica está en el cribado de pacientes con grados más leves del SHP (ver arriba).
En resumen, no existe un algoritmo único aceptado por todos los grupos de trabajo en cuanto a la estrategia de cribado del SHP y cualquiera de los referidos es válido. Los basados en la pulsioximetría inicial podrían infradiagnosticar los casos leves; no obstante en estos pacientes no se ha demostrado un aumento de mortalidad ni pretrasplante ni postrasplante por lo que la importancia de su diagnóstico, en ausencia de un tratamiento médico eficaz, tendría escasa relevancia clínica. La realización de una gasometría arterial de inicio con determinación del PA-aO2 ≥ 15 (≥ 20 en mayores de 65 años) mmHg en lugar de la pulsioximetría mejoraría la sensibilidad diagnóstica para la detección de hipoxemia arterial. Finalmente, la realización de un ECC como cribado inicial con gasometría arterial en los ECC positivos, tal y como proponen Porres et al.,5 permite descartar en un primer paso el SHP mediante el despistaje de las dilataciones vasculares para posteriormente confirmar el diagnóstico mediante gasometría arterial en los pacientes con ECC positivo (fig. 2). Nosotros hemos seleccionado este algoritmo ya que, además de lo expuesto, permite conocer la evolución natural de los pacientes que presentan dilataciones vasculares sin alteraciones gasométricas.
Diagnóstico diferencialEn hasta un 30% de los pacientes con SHP se dan otras enfermedades pulmonares como la enfermedad pulmonar obstructiva crónica o la fibrosis pulmonar que contribuyen a incrementar el grado de hipoxemia, siendo necesario un adecuado juicio clínico para llegar a un correcto diagnóstico. Por otra parte, la propia enfermedad hepática avanzada se asocia con diferentes complicaciones pulmonares45. En este sentido, pueden coexistir alteraciones pleurales o diafragmáticas como el hidrotórax hepático o el efecto de la ascitis masiva sobre la función pulmonar; o alteraciones parenquimatosas como el enfisema panacinar en el déficit de alfa1-antitripsina o la neumonía por aspiración debido a la encefalopatía hepática.
Además del SHP, en la enfermedad hepática se pueden producir otros trastornos de origen vascular que pueden originar hipoxemia y que a menudo se confunden con el SHP. Concretamente, la hipertensión portopulmonar consiste en una obstrucción al flujo en el lecho arterial pulmonar causada por vasoconstricción y proliferación del endotelio vascular y del músculo liso que incrementa las resistencias vasculares pulmonares causando hipoxemia, fallo cardiaco derecho y muerte46. Finalmente, en el diagnóstico diferencial de los trastornos vasculares pulmonares asociados a enfermedades hepáticas también se encontrarían la teleangiectasia hereditaria hemorrágica (síndrome de Rendu-Osler-Weber) y las anastomosis cavopulmonares después de intervenciones por cardiopatías congénitas que pueden remedar un SHP5,17.
Sin embargo, la combinación de una desoxigenación arterial, dilataciones vasculares intrapulmonares y enfermedad hepática es tan particular y único que sostiene el diagnóstico de SHP incluso en presencia de enfermedades crónicas cardiorrespiratorias como la enfermedad pulmonar obstructiva crónica, asma bronquial o la fibrosis idiopática pulmonar, las cuales pueden también causar o agravar las alteraciones en el intercambio de gases2. No obstante, en presencia de una enfermedad pulmonar concomitante y de una desoxigenación puesta de manifiesto por una alteración del PA-aO2 o una hipoxemia, dilataciones vasculares y enfermedad hepática, se puede recurrir a la realización de una γMA con el fin de determinar si la alteración gasométrica es debida al SHP5; esta prueba permite además cuantificar el shunt intrapulmonar y dar información de cara al pronóstico. En la tabla 2 se resumen el diagnóstico diferencial de la disnea en pacientes cirróticos.
Diagnóstico diferencial de la disnea en pacientes con cirrosis
| Enfermedades cardiopulmonares intrínsecas |
| Fallo cardíaco congestivo |
| Enfermedad pulmonar obstructiva crónica (EPOC)/asma/enfermedades pulmonares restrictivas |
| Complicaciones de la cirrosis/hipertensión portal (HTP) |
| Ascitis |
| Hidrotórax hepático |
| Anemia |
| Debilidad muscular |
| Complicaciones pulmonares de enfermedades específicas |
| Déficit Alfa 1 antitripsina: enfisema panacinar |
| Fibrosis quística: bronquiectasias |
| Cirrosis biliar primaria: alveolitis fibrosante, hemorragia y/o granulomas pulmonares |
| Sarcoidosis: enfermedad pulmonar intersticial, hipertensión pulmonar |
| Complicaciones vasculares pulmonares de la cirrosis |
| Síndrome hepatopulmonar |
| Hipertensión portopulmonar |
Además de empeorar la calidad de vida, se ha observado que el SHP conlleva un incremento de la mortalidad, salvo en los casos leves y moderados, cuando se realiza un cribado sistemático en los candidatos a TH20–23. Se han comunicado tasas de mortalidad a 2,5 años de 40-60%, con un incremento de la mortalidad de más del doble frente a pacientes sin SHP tras ajustar por el MELD y TH5. El grado de hipoxemia, que se ha estimado progresa a un ritmo de 5mmHg/año, se relaciona con la mortalidad21. Aunque se ha observado un aumento de la mortalidad por accidente cerebrovascular, probablemente relacionado con embolismo paradójico, no existen causas específicas de mortalidad ligadas al SHP que sean independientes de la progresión de la enfermedad hepática y las complicaciones de la HTP5–7.
En la actualidad no existe un tratamiento médico efectivo para el SHP siendo el TH el único tratamiento eficaz5–7. En cuanto a la terapia farmacológica, se han comunicado pequeños estudios no controlados utilizando distintas clases de fármacos dirigidos a distintos mecanismos implicados en la fisiopatogenia tanto de la HTP como del propio SHP. Así, se han comunicado casos aislados o pequeñas series de pacientes tratados con análogos de somatostatina, beta-bloqueantes, inhibidores de la ciclooxigenasa, indometacina, glucocorticoides, inmunosupresores como la ciclofosfamida, mofetil micofenolato (con acción inhibidora de la angiogénesis y de la producción de ON), pentoxifilina (con acción anti-TNF), inhibidores del ON, ON inhalado, vasoconstrictores pulmonares (almitrina), antibióticos para reducir la translocación bacteriana intestinal2,47–62. Ninguno de estos estudios ha podido demostrar eficacia en cuanto a la mejoría en la oxigenación ni en la vasodilatación pulmonar probablemente influenciado por el diseño de los estudios y su pequeño tamaño muestral. También se ha estudiado el ajo, al cual se atribuyen propiedades vasodilatadoras que pueden redistribuir el flujo pulmonar mejorando el desequilibrio V/Q. Concretamente, en un ensayo clínico doble-ciego, controlado con placebo, las cápsulas de aceite de ajo se asociaron con una mejoría significativa de la oxigenación junto a una resolución del SHP en el 67 frente al 5% con placebo63; sin embargo, el estudio se realizó sobre un pequeño número de pacientes (20 pacientes por brazo) y el grupo placebo presentó una mejoría de 5mmHg al año en lugar de una esperada reducción de esa misma magnitud. Por todo ello, se precisan ensayos clínicos aleatorizados y controlados con placebo, bien diseñados y con suficiente potencia estadística para investigar estas u otras nuevas dianas terapéuticas en esta patología.
En cuanto a las medidas no farmacológicas, la oxigenoterapia mejora la clínica de estos pacientes y debe administrarse cuando la PaO2 cae por debajo de 60mmHg ya que la hipoxemia crónica per se puede contribuir a la mortalidad por SHP6. La utilización de una comunicación portosistémica intrahepática transyugular (TIPS en sus siglas en inglés) en el SHP se ha comunicado en algunas series habiendo obtenido resultados discordantes, por lo que debe considerarse aún una técnica en investigación sin que puedan establecerse recomendaciones en la actualidad basadas en los datos limitados disponibles6,64.
En la actualidad, el TH representa la mejor opción terapéutica para los pacientes con SHP e hipoxemia severa20–22. Según los datos de un estudio reciente, en estos pacientes la supervivencia en lista de espera de TH no difiere de la supervivencia de los pacientes cirróticos sin SHP aunque estos resultados pueden estar influenciados por la gravedad de los casos incluidos y el tiempo medio de espera en lista23. La mortalidad en los primeros 6 meses pos-TH, en pacientes con SHP oscila entre 21-33% según diferentes estudios12,21,23,65 no existiendo diferencias en las tasas de mortalidad pos-TH a largo plazo entre pacientes con SHP y sin SHP21,23. El TH es el único método eficaz de tratamiento capaz de revertir permanentemente la hipoxia del SHP y de conducir a la resolución completa del SHP en la mayoría de los supervivientes23,65. El grado de mejoría de la oxigenación arterial es variable, ocurriendo entre meses y años después del TH y variando según el grado de hipoxemia pretrasplante. En este sentido, el trabajo más completo y con mayor número de pacientes publicado hasta la fecha23, observó que las anormalidades del SHP generalmente se resuelven en más del 95% de los pacientes a los 6 meses postrasplante y en el 100% de los pacientes al año. El único parámetro que se ha observado que puede persistir alterado más allá del año, e incluso, que puede no corregirse a largo plazo tras el TH, es la DLCO23,66, aunque no existe una teoría demostrada que explique esta anormalidad.
Debido al empeoramiento del pronóstico originado por el SHP y su respuesta al trasplante, en EE. UU. estos pacientes se priorizan mediante el sistema MELD según el grado de hipoxemia recibiendo puntos añadidos. Así, desde 2007, la United Network for Organ Sharing estableció una priorización con una puntuación de 22 para los pacientes con SHP e hipoxemia severa (PaO2<60mmHg) añadiendo puntos cada 3 meses6,7. En el estudio retrospectivo más amplio que ha analizado los resultados de esta actuación se observó una mejoría de la supervivencia a los 5 años por esta indicación, del 67% en la era pre-MELD hasta el 88% en la era pos-MELD, siendo esta última similar a la alcanzada para otras indicaciones en la era pos-MELD67. Sin embargo, los resultados de un reciente estudio retrospectivo que evalúa el impacto de las políticas de priorización en este tipo de pacientes cuestionan esta política que podría priorizar de manera excesiva a los pacientes con SHP frente a los que no tienen SHP por lo que está siendo revisada por la United Network for Organ Sharing68. La Sociedad Española de Trasplante Hepático ha difundido el pasado año un documento de consenso sobre las excepciones al modelo MELD en la priorización del trasplante hepático considerando al SHP dentro de estas excepciones69. Concretamente, el grupo de consenso considera que hay datos suficientes para justificar prioridad adicional en pacientes con PaO2<60mmHg, shunt intrapulmonar demostrado y sin otras causas de hipoxemia. Por otra parte, la hipoxemia es un criterio adecuado para determinar la progresión de la enfermedad en lista de espera, ya que es una variable específica, objetiva y fácil de obtener70. En este contexto, el Grupo de Consenso considera que una PaO2<50mmHg parece un límite adecuado para considerar la exclusión de lista por su alta repercusión sobre la supervivencia postrasplante. Según el algoritmo de la Task Force on Pulmonary-Hepatic Vascular Disorders, si la hipoxemia es muy grave (PaO2<50mmHg) y/o existe comorbilidad cardiopulmonar, el TH se debe considerar de forma individualizada tras una completa valoración de la severidad y el pronóstico de las condiciones asociadas5.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Fórmula abreviada del cálculo del gradiente alveolo-arterial de oxígeno (PA-aO2): PAO2 –PaO2 = (FiO2 [Patm-PH2O] – [PaCO2/0,8]) – PaO2 donde PAO2 es la presión parcial alveolar de oxígeno, PaO2 es la presión parcial arterial de oxígeno, FiO2 es la fracción de oxígeno inspirado (0,21 respirando aire ambiente), Patm es la presión atmosférica (760 mmHg al nivel del mar), PH2O es la presión parcial del vapor de agua a la temperatura corporal (47 mmHg a 37°C), y PaCO2 es la presión parcial arterial de dióxido de carbono; 0,8 es la razón estándar de intercambio gaseoso respiratorio en reposo. El rango normal es de 4 a 8 mmHg. El rango normal para la presión parcial de oxígeno es de 80 a 100 mmHg al nivel del mar, mientras el paciente respira aire ambiente en reposo. Para pacientes mayores de 65 años, se utiliza un valor de ≤ 70 mm Hg para PaO2 y un valor de ≥ 20 mmHg para el gradiente alveolo-arterial de oxígeno.

