




Sr. Director: El síndrome de Sweet, descrito por Robert Douglas Sweet1 en 1964, se caracteriza por la aparición de placas cutáneas eritematosas y dolorosas, fiebre y leucocitosis. Éstas se localizan sobre todo en las extremidades superiores, la cara y el cuello. Las lesiones suelen presentarse como nódulos o pápulas de color rojo-violeta, que forman placas grandes con bordes irregulares, o como lesiones pustulosas que, en la mayoría de los casos asientan en las manos2. Histológicamente, se observa un edema papilar con gran infiltrado de polimorfonucleares, en ausencia de vasculitis y necrosis dérmica.
Su incidencia es de 3 casos por 106/habitantes/año3. El síndrome se asocia, en la mitad de los pacientes, a leucemias, conectivopatías, enfermedad de Crohn, colitis ulcerosa y otros procesos, por lo que se considera un marcador cutáneo de enfermedad sistémica4. Se presenta el caso de una mujer con colitis ulcerosa, artritis de tobillos y síndrome de Sweet.
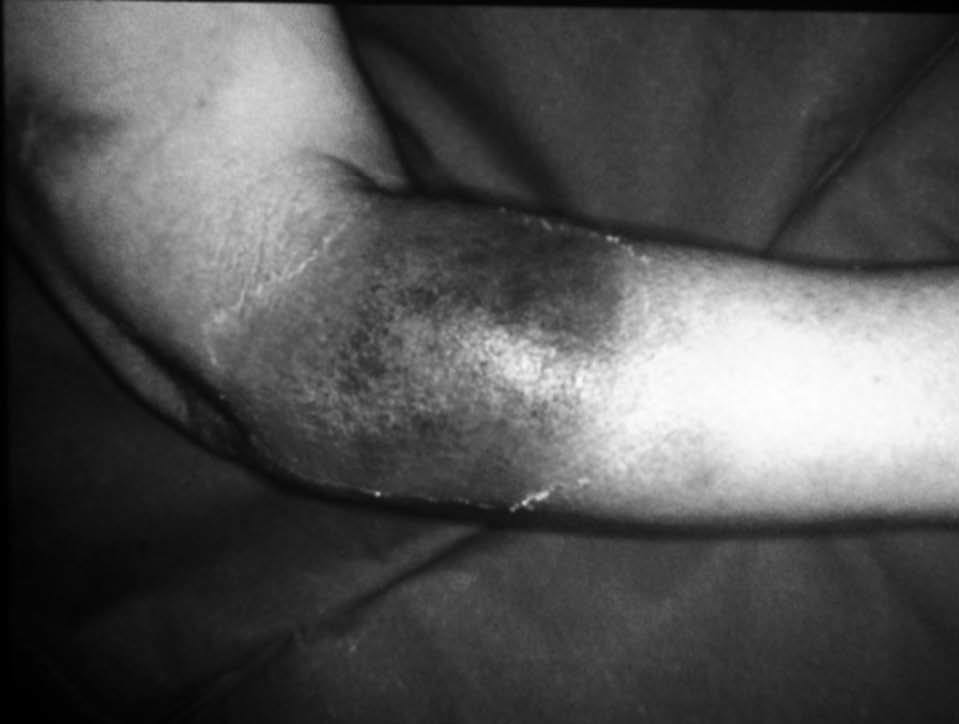
Mujer de 35 años de edad, en tratamiento con anticonceptivos orales, sin antecedentes personales de interés, y madre con artritis reumatoide seropositiva. Desde hace 6 meses refería una alteración del hábito intestinal, con deposiciones diarreicas ocasionales con sangre y, esporádicamente, tenesmo rectal e incontinencia. No presentaba síndrome constitucional. Desde hace un mes, también presentaba artritis en las rodillas y, posteriormente, en los tobillos. Desde hace 3 días presentaba aftas en la boca y en el aparato genital, y tenía una fiebre de 38 °C, por lo que acudió al hospital. En la exploración física destacaban, además de las aftas, una erupción maculopapulosa, dolorosa en la región anterior de los muslos y los brazos (fig. 1A), junto con tumefacción, aumento de la temperatura local y derrame articular en ambos tobillos.
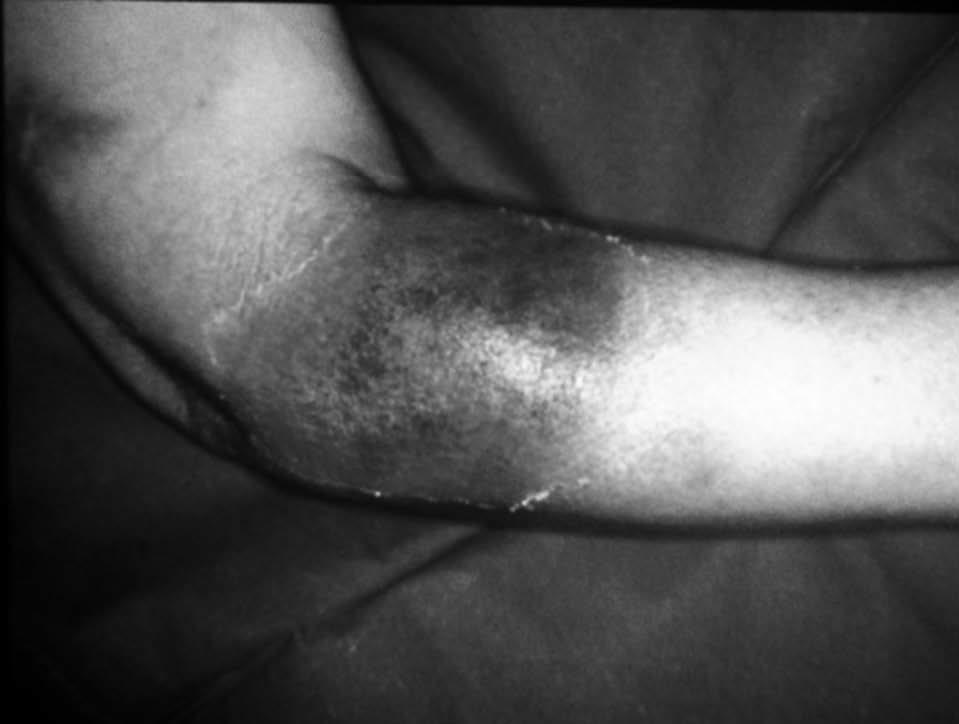
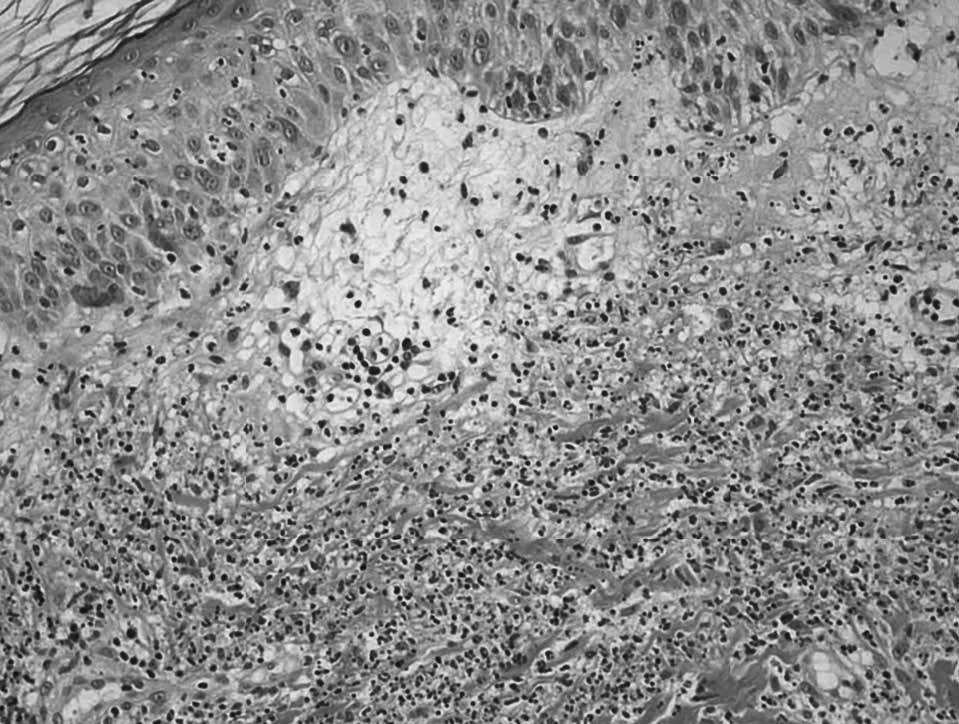
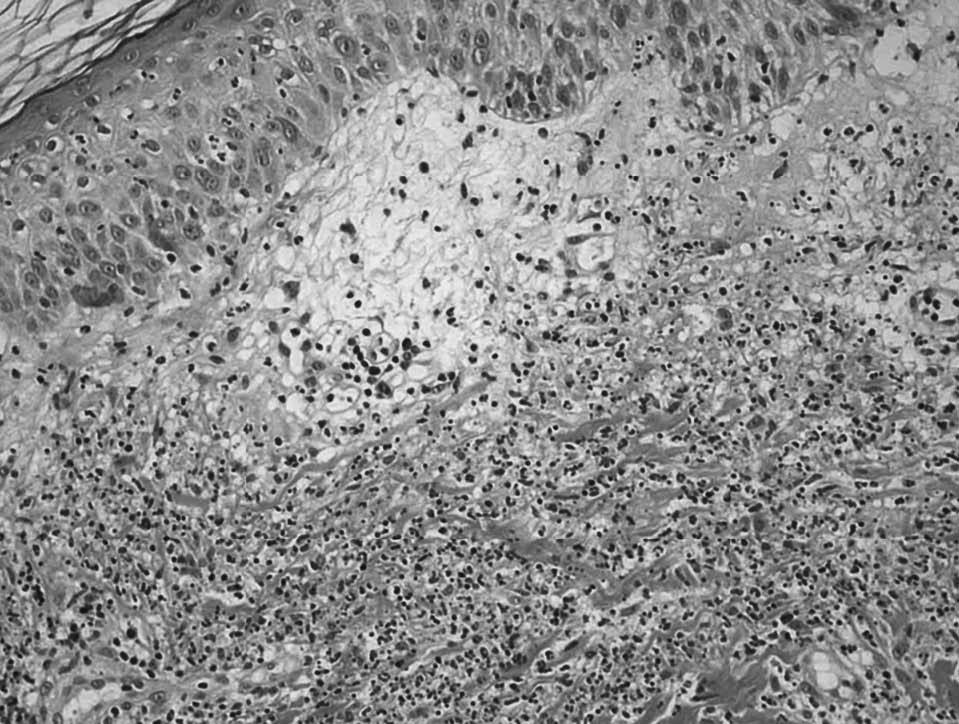
Fig. 1. A) Lesión papulosa (placa) en paciente con colitis ulcerosa. B) Edema en dermis papilar y denso infiltrado leucocitario con leucocitoclaxis, sin vasculitis.
En los datos analíticos se confirmaba anemia (hemoglobina 10,2 g/dl, hematocrito 30,7%), leucocitosis con neutrofilia (14.250 leucocitos/µl; 89% de neutrófilos), velocidad de sedimentación corpuscular de 60 mm/h y proteína C reactiva de 97 mg/dl. Se realizó una sigmoidoscopia de los últimos 60 cm del colon, en la que se observó una mucosa friable, edematosa y granujienta, con pequeñas ulceraciones. Las biopsias del colon fueron compatibles con una colitis ulcerosa. Se biopsiaron las lesiones que presentaba en los muslos, confirmándose el diagnóstico clínico de síndrome de Sweet (fig. 1B). Ante estos hallazgos, se inició tratamiento con 60 mg de prednisona intravenosa al día. Al persistir con 8-10 deposiciones sanguinolientas diarias, se añadió azatioprina 50 mg/día, y tras recibir tiopurina S-metil transferasa se incrementó a 75 mg/día. Al alta, la paciente se encontraba afebril, sin dolor articular ni flogosis, el número de deposiciones diarias disminuyó a 3 y habían mejorado sus lesiones dérmicas.
La asociación del síndrome de Sweet con la colitis ulcerosa fue descrita por primera vez por Benton5, en 1985. Tras la búsqueda de los casos con esta asociación en diversas bases de datos UpToDate, Clinical Evidence, Infopoems, Trip, Cochrane y PubMed, hemos encontrado aproximadamente 20 casos hasta el año 2007. Algunos autores han sugerido como mecanismo etiopatogénico del síndrome la infección, ya que en varias ocasiones se han hallado cultivos y serologías positivos para Yersinia enterocolítica5,6. En la actualidad se ha comprobado que hay una alteración de la función de los neutrófilos y de la regulación de citocinas e interlecucinas 1, 3, 6 y 87.
El presente caso reúne las características clínicas propias de la asociación de síndrome de Sweet y de enfermedad inflamatoria intestinal. Éstas fueron estudiadas por Travis et al6 en 1997, tras revisar 30 casos (20 de enfermedad de Crohn y 10 de colitis ulcerosa), y son las siguientes: sexo femenino (87% de los pacientes), localización de la enfermedad inflamatoria intestinal en el colon (100%), actividad de la colitis ulcerosa (67%) y presencia de otras manifestaciones extraintestinales (77%). El tratamiento con corticoides mejora las lesiones dérmicas y también los síntomas digestivos. Se recomienda iniciar la terapia en dosis altas (1 mg/kg/día), mantener la prednisona durante varias semanas y disminuirla, de forma paulatina, a durante 3-5 meses. Se han utilizado otras alternativas (dapsona, colchicina, clofazimina, ciclosporina y doxiciclina) con resultado variable8.

