




The most common hereditary gastrointestinal cancers are colorectal, mainly hereditary nonpolyposis colorectal cancer (Lynch syndrome) and familial adenomatous polyposis. Other extracolonic neoplasms, including the gastric and pancreatic adenocarcinomas, are less well known and studied because they account for a relatively small percentage of hereditary gastrointestinal cancers. Nonetheless, they merit special attention because of the high associated morbidity and mortality rates. We review the hereditary and familial syndromes associated with gastric and pancreatic cancers with a view to improving knowledge and understanding of these diseases, in order to heighten diagnostic suspicion and thus implement appropriate diagnostic strategies, screening, surveillance and treatment.
De todas las neoplasias digestivas hereditarias, las más importantes por su frecuencia son las que afectan al colon y recto, principalmente el síndrome de Lynch y la poliposis adenomatosa familiar. Sin embargo existen otros grupos de neoplasias digestivas extracolónicas muy poco estudiadas y conocidas, que constituyen un pequeño porcentaje de los cánceres hereditarios digestivos. A pesar de ser poco frecuentes, estas neoplasias merecen importancia debido a la gran morbimortalidad que conllevan, destacando principalmente el adenocarcinoma gástrico y pancreático. Este artículo tiene como objetivo hacer una revisión de los datos conocidos hasta la fecha de los síndromes hereditarios y familiares asociados a estas dos neoplasias, de cara a un mayor conocimiento y entendimiento de estas patologías, con la intención de mejorar la sospecha diagnóstica y así poner en marcha las estrategias diagnósticas, de cribado, de vigilancia y terapéuticas adecuadas.
Gastric cancer (GC) is the fifth most common cancer worldwide, and one of the leading causes of cancer-related death. The incidence in Spain is 7.8 cases per 100,000 population, and it is twice as common in men than in women.1 Mean age at diagnosis is 60 years. Only 7% of patients present before age 50, and 2% before age 40.2
The aetiology of GC is multifactorial, with the main environmental agents including Helicobacter pylori (H. pylori), diet and smoking. Histologically, it is classified mainly into intestinal- or diffuse-type adenocarcinoma. Intestinal GC is more closely associated with environmental factors and old age, whilst diffuse GC occurs in younger people and is characterised by a multifocal signet ring cell infiltrate.2
Although most GC are sporadic, familial aggregation can be observed in approximately 10% of cases.2,3 Hereditary GC, i.e. in the context of an associated germline mutation, accounts for 1%–5% of all GCs.4 Family characteristics that suggest a hereditary predisposition include the existence of several affected family members, an autosomal dominant pattern of inheritance, disease presentation at young ages and association with other extragastric neoplasms.
Based on these parameters, there are 3 clinical situations in which familial predisposition to GC may be found:
- 1)
Hereditary syndromes with higher risk mainly of GC. This scenario includes two entities:
Hereditary diffuse gastric cancer (HDGC), and 1.2 Gastric adenocarcinoma and proximal polyposis of the stomach (GAPPS).
- 2)
Hereditary syndromes with higher risk of GC and other tumours: in the context of other hereditary cancer syndromes that are associated with a higher risk of GC and other tumours, with an associated germline mutation (Table 1).
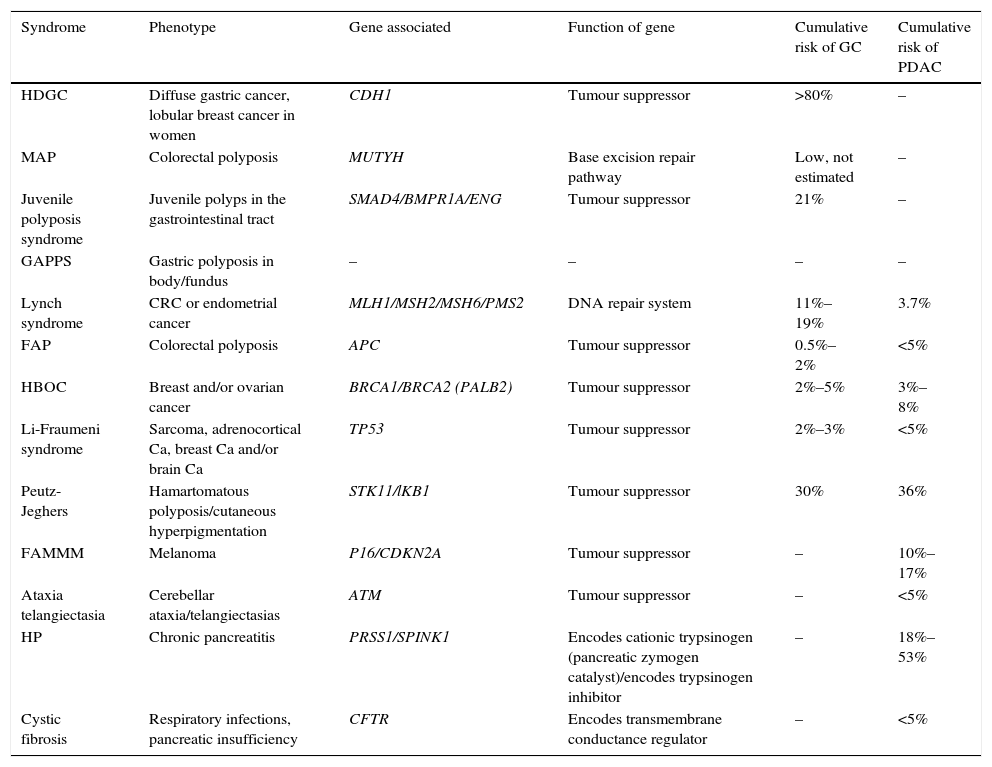
Table 1.Hereditary cancer syndromes associated with gastric adenocarcinoma and pancreatic ductal adenocarcinoma.
Syndrome Phenotype Gene associated Function of gene Cumulative risk of GC Cumulative risk of PDAC HDGC Diffuse gastric cancer, lobular breast cancer in women CDH1 Tumour suppressor >80% – MAP Colorectal polyposis MUTYH Base excision repair pathway Low, not estimated – Juvenile polyposis syndrome Juvenile polyps in the gastrointestinal tract SMAD4/BMPR1A/ENG Tumour suppressor 21% – GAPPS Gastric polyposis in body/fundus – – – – Lynch syndrome CRC or endometrial cancer MLH1/MSH2/MSH6/PMS2 DNA repair system 11%–19% 3.7% FAP Colorectal polyposis APC Tumour suppressor 0.5%–2% <5% HBOC Breast and/or ovarian cancer BRCA1/BRCA2 (PALB2) Tumour suppressor 2%–5% 3%–8% Li-Fraumeni syndrome Sarcoma, adrenocortical Ca, breast Ca and/or brain Ca TP53 Tumour suppressor 2%–3% <5% Peutz-Jeghers Hamartomatous polyposis/cutaneous hyperpigmentation STK11/lKB1 Tumour suppressor 30% 36% FAMMM Melanoma P16/CDKN2A Tumour suppressor – 10%–17% Ataxia telangiectasia Cerebellar ataxia/telangiectasias ATM Tumour suppressor – <5% HP Chronic pancreatitis PRSS1/SPINK1 Encodes cationic trypsinogen (pancreatic zymogen catalyst)/encodes trypsinogen inhibitor – 18%–53% Cystic fibrosis Respiratory infections, pancreatic insufficiency CFTR Encodes transmembrane conductance regulator – <5% Ca: cancer; CRC: colorectal cancer; FAMMM: familial atypical multiple mole melanoma; FAP: familial adenomatous polyposis; GAPPS: gastric adenocarcinoma associated with proximal polyposis of the stomach; HBOC: hereditary breast and ovarian cancer syndrome; HDGC: hereditary diffuse gastric cancer; HP: hereditary pancreatitis; MAP: MUTYH-associated adenomatous polyposis.
- 3)
Familial gastric cancer: situation in which a familial aggregation of GC of intestinal histology is observed with no identified genetic cause.
This entity accounts for between 1% and 3% of all GCs. It was first described in 1998 following the identification of a germline mutation in the CDH1 gene in families of Maori origin with multiple cases of diffuse GC.5 Since then, CDH1 mutations have been identified in up to 50% of families meeting clinical criteria for HDGC. A recently published study6 that analysed the largest reported series of CDH1 mutation carriers to date identified a 70% cumulative risk of GC by the age of 80 years for male CDH1 mutation carriers, and a 56% cumulative risk for females, as well as a 42% cumulative risk of breast cancer for females by the age of 80 years. The study also established a potential increased risk of colorectal and prostatic cancer.7,8
Genetics. Autosomal dominant inheritance pattern. HDGC is associated with germline mutations in the CDH1 gene in 30%–50% of cases. This gene is located on chromosome 16q22.1 and encodes the protein E-cadherin. It acts as a tumour suppressor gene, with mutation leading to loss of cell adhesion, increasing proliferation, invasion and metastasis. In individuals with a CDH1 germline mutation, somatic inactivation of the healthy CDH1 allele occurs, predisposing the individual to the formation of tumours in the locations described.
More than 100 CDH1 germline mutations have been identified. Most mutations are truncating (mainly frameshift mutations), splice-site and nonsense mutations, followed by mutations in which one amino acid is changed for another (missense mutations).9,10 Mutations in the alpha-E-catenin gene (CTNNA1) have also been identified as causative of HDGC.11
Penetrance is high, although variable–an estimated 63%–83% in women and 40%–67% in men.
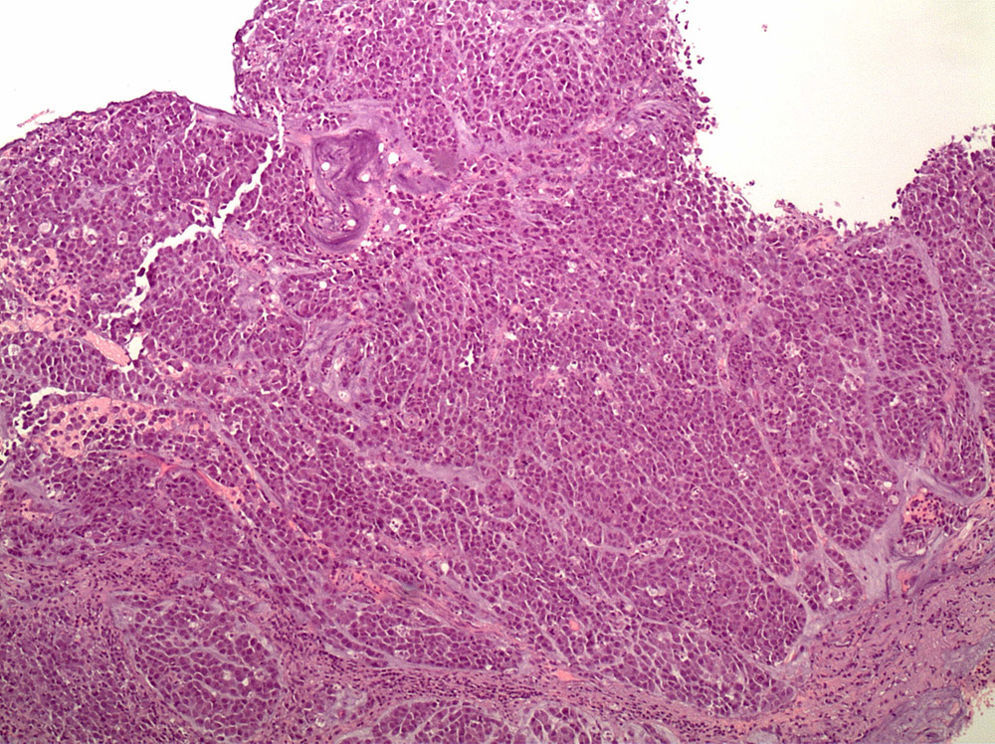
Histopathology. These tumours are characterised by diffuse histology with signet ring cells (Fig. 1); the tumour foci can be multiple and spread throughout the gastric mucosa, with a higher concentration in the antrum and body-antral transitional zone.
.")
Diagnostic criteria. According to the 2004 guidelines of the International Gastric Cancer Linkage Consortium, updated in 2015, the criteria that define HDGC–and therefore the situations in which germline genetic testing (study of mutations in the CDH1 gene) is indicated–are as follows12:
- •
Family with ≥2 cases of diffuse gastric cancer (DGC), with at least 1 confirmed case, in first degree relatives (FDR) or second degree relatives (SDR) regardless of age at diagnosis, or
- •
Individuals with DGC diagnosed before the age of 40 with no family history, or
- •
Personal or family history of DGC and lobular breast cancer (LBC), with 1 of these diagnosed before the age of 50.
A histopathology report–preferably assessed by a pathologist experienced in GC–is essential to properly evaluate whether a family meets the criteria for HDGC. Confirmed cases of intestinal-type GC do not form part of HDGC; testing for CDH1 mutations is therefore not indicated in these families.
Genetic testing should also be considered in the following families12:
- •
Bilateral LBC or family history of ≥2 cases of LBC <50 years old.
- •
Personal or family history of cleft lip/palate in a patient with DGC.
- •
In situ signet ring cells and/or spread of signet ring cells.
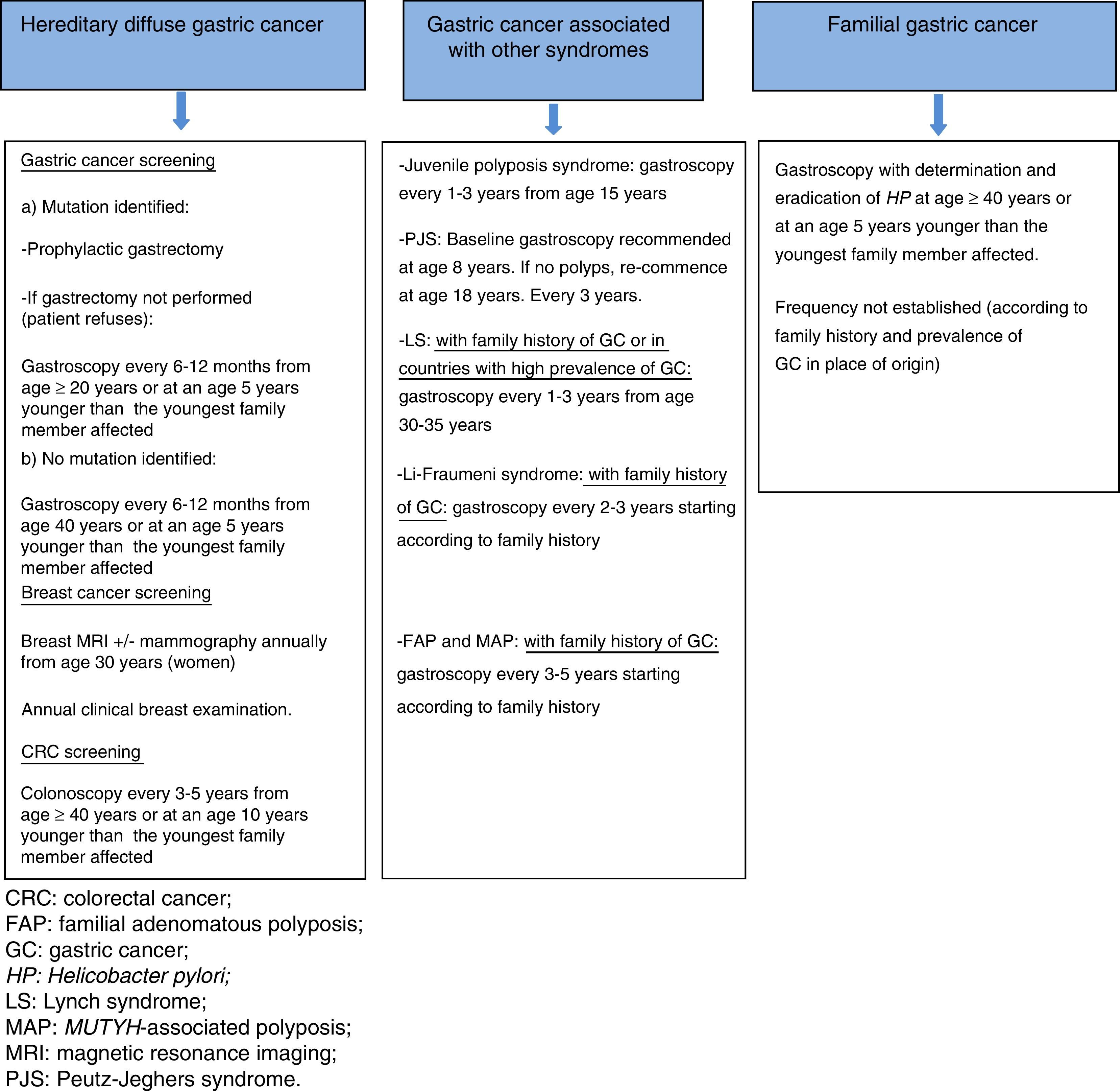
*Prophylactic gastrectomy. In most asymptomatic patients who are carriers of a germline mutation in CDH1, macroscopic lesions are not found on endoscopic examination; however, intramucosal foci of DGC, generally multiple, can be seen in the surgical specimens.13 Prophylactic total gastrectomy is therefore advised in carriers of a pathogenic mutation who are aged over 20 years, recommending surgery at an age 5 years younger than the youngest family member with GC (the mean age at DGC diagnosis is 38–40 years).7,14
*Endoscopic screening. This is reserved for patients who do not opt for prophylactic gastrectomy, patients with a variant of uncertain significance, and patients in whom the germline mutation could not be identified. It is established in accordance with the Cambridge protocol.14
- •
High definition endoscopy, careful inspection for 30min, gastric inflation/deflation manoeuvres, use of mucolytics, biopsy of all mucosal abnormalities, serial biopsies: 30 random biopsies are recommended, 6 from each region (antrum, incisura, body, fundus and cardia), and detection and eradication of H. pylori.
Endoscopic screening is recommended every 6–12 months, starting at age 20, or at an age 5 years younger than the youngest family member with GC in patients with a known mutation and who refuse gastrectomy, and from 40 years or at an age 5 years younger than the youngest family member with GC in cases with no identified pathogenic mutation12,14 (Fig. 2).

Other neoplasms. Annual breast magnetic resonance imaging (MRI) (which can be combined with mammography) is recommended from age 30 in female carriers of a CDH1 germline mutation.12 Annual clinical breast examination is also essential. In families with a known mutation and cases of colorectal cancer (CRC), colonoscopy is recommended every 3–5 years from age 40 or at an age 10 years younger than the youngest family member affected.12
Gastric adenocarcinoma and proximal polyposis of the stomachThis is a recently described syndrome of proximal gastric polyposis with a high risk of intestinal GC. The first family identified was in Australia, with other families subsequently identified in the United States and Canada. Patients typically present fundic gland polyposis with areas of dysplasia or intestinal-type gastric adenocarcinoma, restricted to the proximal stomach, with no evidence of duodenal or colonic polyposis. The age at presentation of GC is variable (cases have been identified from 33 to 75 years), with an anticipation effect.4,15
Genetics. Autosomal dominant inheritance. Incomplete penetrance. The causal genetic defect has not been identified.
Histopathology. Characterised by florid gastric polyposis, generally >100 polyps measuring <10mm in diameter, which carpet the gastric body and fundus. The oesophagus, gastric antrum, pylorus and duodenum are generally unaffected. Histology is consistent with fundic gland polyposis, including areas of dysplasia and occasionally hyperplastic and adenomatous polyps. Histological cancer type is intestinal.4,15,16
Diagnostic criteria. Diagnosis is based on the presence of the following15:
- •
Gastric polyposis of the body and fundus with no evidence of colorectal or duodenal polyposis and >100 polyps carpeting the proximal stomach in the index case or >30 polyps in an FDR of a patient with GAPPS and predominantly fundic polyps, some with dysplasia and a family member with dysplastic fundic polyps or GC and
- •
Autosomal dominant pattern.
This syndrome must be distinguished from hereditary syndromes, such as classic or attenuated familial adenomatous polyposis (FAP), MUYTH-associated polyposis (MAP) and Peutz-Jeghers syndrome (PJS). The use of proton pump inhibitors also forms part of the differential diagnosis; accordingly, these must be excluded in order to make a diagnosis of GAPPS and the endoscopy repeated after discontinuing proton pump inhibitor therapy to rule out any association.
GC prevention and screening. Since few families have been identified to date, management is not well established. The risks and benefits of prophylactic gastrectomy and endoscopic screening should be assessed on an individual basis, taking into consideration the limitations of endoscopic surveillance in each case and the risk of GC in each family. Gastroscopy and colonoscopy are recommended in FDRs of affected individuals.4
Hereditary syndromes with higher risk of gastric cancer and other tumoursGC can present in the context of other cancer predisposition syndromes, such as Lynch syndrome (LS), Li-Fraumeni syndrome (LFS), PJS, FAP, MAP. juvenile polyposis syndrome and hereditary breast and ovarian cancer syndrome (HBOC)17 (Table 1).
Lynch syndrome (hereditary nonpolyposis colorectal cancer)LS is associated mainly with an increased risk of CRC, although it also carries a risk of other tumours. Neoplasms on the Lynch spectrum include endometrial cancer, and also ovarian, stomach, bile duct, small bowel, pancreatic, ureter and renal pelvis cancers, as well as skin cancer (sebaceous tumours, in the variant known as Muir-Torre syndrome) and central nervous system tumours (glioblastomas and astrocytomas, in the variant known as Turcot syndrome). The risk of GC in this context is estimated to be between 6% and 13%, with a mean age at diagnosis of 56 years.18,19
Genetics. This syndrome has autosomal dominant inheritance, caused by germline mutations in one of the genes of the DNA repair system (MLH1, MSH2, MSH6 or PMS2). MLH1 is located on chromosome 3p21, MSH2 on chromosome 2p16, MSH6 on chromosome 2p16 and PMS2 on chromosome 7p22. The role of DNA repair genes is to maintain the integrity of the genome by correcting base substitution errors and small insertions-deletions that are generated during DNA replication, and which can affect regulation of cell growth and promote formation of neoplasms.20
Histopathology. In the context of LS, GC is intestinal in 90% of cases, with only a small percentage presenting diffuse histology. Germline mutations in the DNA repair genes give rise to 2 phenomena detectable within the tumour: the presence of microsatellite instability (MSI), and loss of expression of the protein corresponding to the affected gene (detectable by immunohistochemistry).
Diagnostic criteria. The Amsterdam criteria, described in 1991, were key for defining this syndrome, although they are very strict (they are only met by <40% of families) and initially did not consider GC. The revised Bethesda guidelines were later developed, and are probably the criteria most widely used to determine which CRC patients should undergo study of the DNA repair system in the tumour, in order to focus germline genetic testing on those with abnormalities in this system (presence of MSI and/or loss of expression of MLH1/MSH2/MSH6/PMS2 proteins identified by immunohistochemistry). In parallel to the development of clinical criteria, different mathematical models have been established in recent years. Their aim is to facilitate the identification of patients with LS by predicting the risk of being a carrier of a mutation in one of the DNA repair genes, based on the individual's personal and family history of cancer. Currently, the main models are MMRpro, MMRpredict and PREMM.1,2,6 Although these have been shown to provide satisfactory sensitivity and specificity, and also quantify the risk of being a mutation carrier, clinical suspicion and a complete and accurate family history are needed, so they are mainly used in centres that evaluate high-risk patients.21,22
GC screening. Screening is not well established. Some groups recommend determination and eradication of H. pylori.23
Although there is no solid evidence regarding the advisability of screening gastroscopy in patients with LS, endoscopic screening is recommended in countries with a high prevalence of this neoplasm, and in families with LS with at least 1 family member with GC. Frequency: every 1–3 years, from age 30 to 35 years.23
Li-Fraumeni syndromeThis is a syndrome with low prevalence that carries an increased risk of multiple primary tumours. Fifty percent of the individuals who carry a germline mutation in the TP53 gene develop a cancer associated with this syndrome before 30 years of age. The 4 main cancers are breast cancer, brain cancer, adrenocortical cancer and sarcoma (accounting for 80% of cases). Other less common neoplasms include leukaemia, lung cancer, melanoma, pancreatic cancer and gastric cancer. The frequency of GC in this syndrome is 2.8%.24,25
Genetics. Autosomal dominant inheritance caused by a germline mutation in the TP53 gene located on chromosome 17p13. TP53 is a tumour suppressor gene that mainly regulates cells with damaged DNA; when the p53 protein malfunctions, the cells with damaged DNA can survive and proliferate, contributing to the malignant transformation.
Histopathology. Tumour histology can be intestinal (70% of cases) or diffuse (30% of cases).
Diagnostic criteria. A mutation in TP53 should be suspected in an individual with any of the following 3 diagnostic criteria (2009 Chompret criteria)26:
- •
Individual with:
- ∘
Tumour belonging to the LFS spectrum diagnosed at <46 years (soft tissue sarcoma, osteosarcoma, premenopausal breast cancer, brain tumour, adrenocortical carcinoma, leukaemia or bronchoalveolar cancer) and,
- ∘
At least 1 FDR or SDR with a tumour belonging to the LFS spectrum (except breast cancer, if the proband has had breast cancer) <56 years of age or with multiple tumours.
- ∘
- •
Individual with multiple tumours (except multiple breast tumours), 2 of which belong to the LFS spectrum, any of which are diagnosed at <46 years.
- •
Individual with adrenocortical carcinoma or choroid plexus tumour, regardless of family history.
GC screening. Screening is indicated if there are cases of GC in the family. Frequency: every 2–3 years, the age of onset is established based on the age at presentation of the GC in the affected family member.27
Peutz-Jeghers syndromeThis syndrome is characterised by the presence of hamartomatous polyps along the gastrointestinal tract and mucocutaneous hyperpigmentation. The cumulative risk of developing cancer at 70 years of age is 85%–90%. The most common neoplasms are breast and colon cancer, followed by pancreatic, stomach and ovarian cancer. The estimated risk of GC is 30%, with a mean age at diagnosis of 30–40 years.28,29
Genetics. Autosomal dominant inheritance. A causal germline mutation in the STK11 gene (also known as LKB1), located on chromosome 19p13.3, has been identified in 70% of cases. STK11 is a tumour suppressor gene that apparently regulates cell polarity, which is related with the development of hamartomas and increased risk of cancer.30
Histopathology. The predominant tumour histology is intestinal adenocarcinoma.
Diagnostic criteria. Clinical diagnosis is established with the presence of 2 or more of the following criteria:
- •
Mucocutaneous pigmentation.
- •
Two or more Peutz-Jeghers-type gastrointestinal hamartomas.
- •
Family history of PJS.
GC screening. Baseline examination at 8 years of age is recommended, continuing every 3 years. If there are no polyps, re-commence endoscopic screening at age 18, every 3 years.31
Familial adenomatous polyposisThis syndrome is characterised by the presence of multiple colorectal adenomas. In its classic form (more than 100 adenomas), it has an almost 100% risk of presenting early-onset CRC if prophylactic colectomy is not carried out. It is associated with a wide spectrum of extracolonic tumours, including hepatoblastoma, and duodenal, pancreatic, thyroid, bile duct and brain adenocarcinoma. The estimated risk of GC is 0.5%–2.0%.32
Genetics. Autosomal dominant inheritance. Associated with germline mutations in the APC gene (chromosome 5q21-q22) in more than 50% of cases of polyposis with more than 100 adenomas. The gene encodes a 2843-amino acid protein (310kDa) that is involved in the Wnt signalling pathway. APC is a tumour suppressor gene that works by negatively regulating the oncoprotein β-catenin, which participates in the regulation of gene transcription such as proliferation, differentiation, apoptosis, etc.33 A mutation that truncates the protein is the most common type, although exonic and whole gene deletions can also be found.
Histopathology. The GC in this syndrome is intestinal.
Diagnostic criteria. Diagnostic suspicion is based on 2 main phenotypes: the classic form, characterised by more than 100 adenomas throughout the colon, and the attenuated form, which presents between 10 and 99 adenomas.
GC screening. Reserved for patients with a family history of GC. Gastroscopy is recommended every 3–5 years, although the start age has not been established and is decided according to family history. In most cases, the surveillance gastroscopy strategy is established based on the follow-up of duodenal polyps, performing the first examination at 25–30 years of age. The gastric polyps most frequently identified in FAP are fundic gland polyps, and less commonly, adenomas. Given the low prevalence of GC in FAP, biopsies/polypectomy is only indicated in gastric polyps that show changes suggestive of malignancy, especially those located in the antrum.34,35
MUTYH-associated polyposisThis syndrome is characterised by an attenuated adenomatous polyposis phenotype, with the presence of fewer than 100 adenomas and in some cases extracolonic manifestations similar to FAP. The risk of extracolonic neoplasms seems to be lower than in FAP. The risk of GC is 1%.
Genetics. This is an autosomal recessive syndrome, caused by biallelic germline mutations in the MUTYH gene (chromosome 1p34.3–1p32.1). MUTYH is a base excision repair gene whose proteins are responsible for repairing oxidative DNA damage. Genes mutated as a result of the oxidative damage exert a major effect on the polyposis phenotype, and the inactivation of MUTYH generates genomic instability in the colorectal epithelial cells, thereby increasing the risk of CRC.36 Most mutations involve amino acid substitution (missense). Several studies support the variability of MUTYH mutations based on geographical and ethnic differences. Thus, in Spain, the 2 most prevalent mutations are G396D and Y179 C (which are the most common mutations in the Caucasian population), although other more specific mutations have been reported, such as p.E410GfsX43.37
Histopathology. There is no information on the histological type of GC in this syndrome.
Diagnostic criteria. Suspected in patients with attenuated or classic adenomatous polyposis phenotype with recessive inheritance pattern.
GC screening. As in FAP, screening is reserved for patients with a family history of GC. Gastroscopy is recommended every 3–5 years, although the start age has not been established and is decided according to family history. In most cases, surveillance upper endoscopy is established based on the follow-up of duodenal polyps, performing the first examination at 25–30 years of age.38
Juvenile polyposis syndromeThis syndrome is characterised by multiple hamartomatous polyps in the gastrointestinal tract (mainly in the colon and stomach) and an increased risk of gastrointestinal cancer. It is the most common hamartomatous syndrome, with an incidence of 1 in 100,000 newborns. The cumulative risk of GC is 21%.39
Genetics. Autosomal dominant inheritance. In 50% of cases it is associated with a germline mutation in the SMAD4 gene located on chromosome 18q21.1, in BMPR1A located on chromosome 10q22-23, or in ENG located on chromosome 9q34.1. These genes are related with the transforming growth factor-beta (TGF-beta) signalling pathway.40
Histopathology. Tumour histology may be intestinal or diffuse.
Diagnostic criteria. Characterised by the presence of at least 1 of the following criteria:
- •
Five or more juvenile polyps in the colon.
- •
Multiple juvenile polyps along the gastrointestinal tract.
- •
Any number of juvenile polyps in a patient with a family history of juvenile polyposis syndrome.
GC screening. Gastroscopy every 1–3 years is recommended, beginning at 15 years of age.31
Hereditary breast and ovarian cancer syndromeThis disorder is associated with an increased risk of breast cancer (47%–55% at age 70 years), ovarian cancer (17%–39%) and other types of cancer, including prostate cancer, male breast cancer, melanoma, pancreatic cancer and stomach cancer. The risk of GC is 2.6%–5.5%.41 Based on a meta-analysis, the relative risk of GC is estimated to be 1.69 (95% CI, 1.21–2.38).42
Genetics. Autosomal dominant inheritance. Most cases are due to mutations in the BRCA1 (17q21.31) or BRCA2 (13q13.1) genes and, less frequently, in the PALB2 (16p12.2) gene.43,44BRCA1 and BRCA2 are tumour suppressor genes and have a major role in the response to cell stress through DNA repair processes.45PALB2 is a gene associated with BRCA2 and has a tumour suppressor function involved in maintaining the genomic integrity.46
Histopathology. Tumour histology may be intestinal or diffuse.
Diagnostic criteria. A mutation in BRCA1/BRCA2/PALB2 should be suspected in an individual with any of the following diagnostic criteria:
- •
Breast cancer <40 years.
- •
Breast and ovarian cancer.
- •
Multiple breast cancer (ipsi/contralateral).
- •
Breast cancer in men.
- •
Breast cancer negative for oestrogen, progesterone and HER2/neu receptors (“triple negative”).
- •
Breast and pancreatic cancer.
- •
High-grade papillary serous ovarian cancer.
- •
Breast cancer at any age and:
- ∘
Two or more family members with breast or pancreatic cancer (regardless of age).
- ∘
One or more family members with breast cancer <50 years.
- ∘
One or more family members with ovarian cancer (regardless of age).
- ∘
Three or more family members with breast cancer (regardless of age).
- ∘
Pancreatic cancer in a patient or family member with breast or ovarian cancer.
GC screening. There are no firm recommendations. Some authors suggest gastroscopy every 2–3 years in patients with a family history of GC, although there is no consensus or guidelines to support this recommendation.38
Familial gastric cancerDefined as cases with familial aggregation of GC of intestinal histology, with no identified genetic cause. The FDR of patients with GC are estimated to have a 2–3-fold increased risk of presenting this cancer, compared to the general population.47
Genetics. The causal germline mutation is unknown.
Diagnostic criteria. The criteria that define familial GC are:
>3 FDR or SDR with CG, regardless of age, or
>2 FDR or SDR with GC, at least 1 diagnosed before 50 years of age.
Histopathology. The tumour histology is intestinal (as diffuse histology is one of the diagnostic criteria for HDGC).
GC prevention and screening. Prophylactic gastrectomy is not recommended. The first gastroscopy should be performed from age 40 years or at an age 5 years younger than the youngest family member affected, with detection and eradication of H. pylori.2 There is no consensus on the advisability and frequency of periodic gastroscopies, with some authors recommending an examination every 2–5 years depending on the family history (Fig. 2).3
Pancreatic cancerPancreatic cancer (pancreatic ductal adenocarcinoma [PDAC]) is a rapidly progressive and generally fatal disease. It is the eighth leading cause of death due to cancer in men and the ninth in women worldwide.48 The incidence in Spain is 6.3 cases per 100,000 population.1 The mean age at diagnosis is 71 years old; only 8%–10% present before age 50 years. Although treatments have improved, PDAC has a mean 5-year survival of <5%.49,50
Smoking, high body mass index, excessive alcohol consumption and diabetes mellitus are factors associated with a higher risk of PDAC. Among the risk factors, smoking is the exogenous risk factor most closely associated with the development of PDAC. It is correlated with a 2–3.7-fold increased risk, and reduces the age of onset of the cancer by around 10 years.
A family history of pancreatic cancer has also been related with a greater risk of developing this neoplasm. This suggests a hereditary component, and the potentially important role of genetic factors in the development of this tumour. Approximately 5%–10% of patients with PDAC have a family history of this neoplasm, showing, therefore, a certain degree of familial aggregation. Several germline mutations involved in hereditary PDAC have been identified.51
Although the main causal gene of PDAC has not yet been identified, it is associated with germline mutations that carry a higher risk of this neoplasm. Recent studies have also described the anticipation phenomenon in 59%–85% of families, which implies that younger generations are usually affected 10 years earlier.52 The most common genetic alterations are: BRCA2, PALB2 bound to the BRCA2 protein, ATM, CDKN2A/p16, and less often: BRCA1, APC, MLH1, MSH2, MSH6, PMS2, PRSS1 and STK11.50,53,54
There are 3 clinical situations in which a predisposition to PDAC may be found: 2 included within hereditary pancreatic cancer (HPC; associated germline mutation) and the third defined as familial pancreatic cancer (FPC; with no identified genetic mutation).
- 1)
HPC in the context of hereditary syndromes with higher risk mainly of pancreatic cancer:
1.1 Hereditary pancreatitis (HP).
1.2 Cystic fibrosis.
- 2)
HPC in the context of hereditary syndromes with higher risk of PDAC and other tumours: in the context of other hereditary cancer syndromes that are associated with a higher risk of PDAC and other neoplasms, with an associated germline mutation (Table 1).
- 3)
Familial pancreatic cancer: situation in which familial aggregation of PDAC is observed with no identified genetic cause.
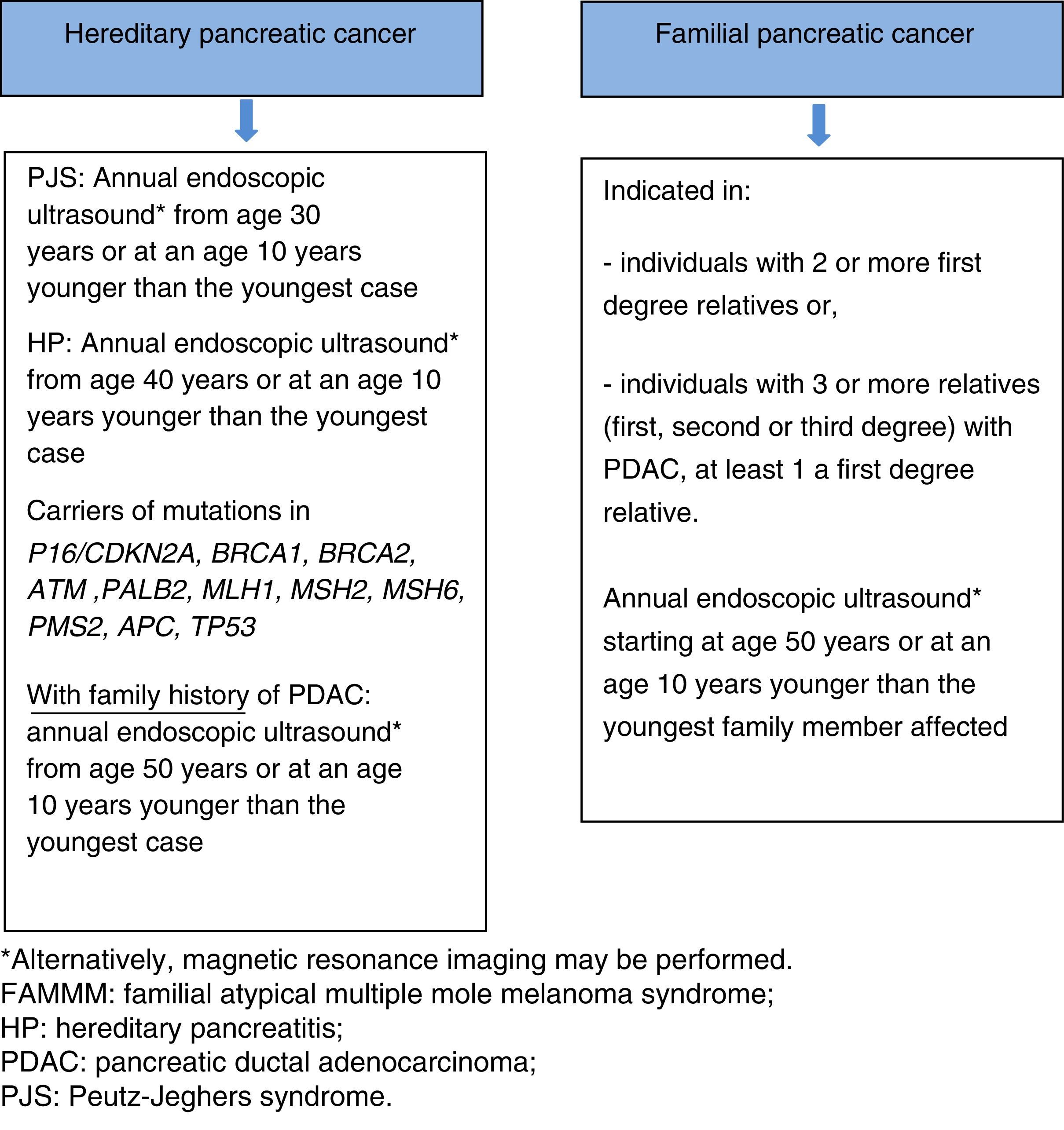
This is a rare disease, and is a hereditary form of chronic pancreatitis. Onset of symptoms begins between the first and second decades of life. Recurrent acute pancreatitis presents that eventually leads to the development of chronic pancreatitis, pancreatic insufficiency, diabetes and risk of PDAC, which varies between 18% and 53% in recent studies.54,55
Genetics. Autosomal dominant inheritance pattern. It is associated with a mutation in the PRSS1 gene (located on chromosome 7q35), with high penetrance (estimated 80%). PRSS1 encodes the protein trypsin 1, which is the main catalyst for the conversion of pancreatic zymogens into pancreatic enzymes. In some cases, HP is associated with the SPINK1 gene (located on chromosome 5q32), which encodes pancreatic secretory trypsin inhibitor.
Diagnostic criteria. Diagnosis is based on the medical history supported by complementary imaging tests and an autosomal dominant pattern of inheritance.
Cystic fibrosisThis is a disease with considerable morbidity and mortality, characterised primarily by persistent respiratory infections, pancreatic insufficiency and high chloride levels in sweat. The risk of PDAC in these patients is not well established, although it is considered to be low (<5%).
Genetics. Autosomal recessive inheritance. It is caused by mutations in the CFTR gene located on chromosome 7q31.2. CFTR encodes the cystic fibrosis transmembrane conductance regulator protein, responsible for regulating chloride channels.
Diagnostic criteria:
- •
Symptoms consistent with cystic fibrosis affecting at least 1 organ and
- •
Cystic fibrosis transmembrane conductance regulator (CFTR) dysfunction:
- ∘
Chloride>60mmol/L in sweat, or
- ∘
Identification of the disease-causing mutation in each CFTR gene, or
- ∘
Abnormal ion transport in the nasal epithelium.
- ∘
HPC can present in the context of other cancer predisposition syndromes, such as HBOC, FAP, LS, PJS, LFS, ataxia telangiectasia and familial atypical multiple mole melanoma syndrome (FAMMM).54,55
Among hereditary syndromes, BRCA2 mutations are the most common form of HPC. The cumulative risk of PDAC in the context of HBOC is 3%–8%. In Spain, the prevalence of PALB2 mutations in families with HBOC, BRCA1/BRCA2-negative, and personal or family history of pancreatic cancer is 1.5%.56 The hereditary syndromes with a higher risk of developing this neoplasm are PJS and FAMMM (with a cumulative risk of 36% and 10%–17%, respectively). PJS, despite being a rare syndrome, is currently the highest known hereditary risk factor for PDAC.
HBOC, PAF, LS, PJS and LFS: already described in the section on GC associated with other hereditary cancer predisposition syndromes (Table 1)Ataxia telangiectasiaThis is a disease associated with a defect in DNA repair mechanisms. Patients with this condition present several neurological abnormalities, such as progressive cerebellar ataxia and abnormal eye movements, as well as ocular and cutaneous telangiectasias, and immune deficiency. This disease has autosomal recessive inheritance (biallelic mutation in the ATM gene), although the increased risk of PDAC is related with heterozygous mutations in the ATM gene. The risk of PDAC in these patients is at least twice that of the general population, accounting for 2.4% of patients with FPC and rising to 4.6% if there are more than 3 cases of PDAC in the family.57
Genetics. Mutation of the ATM gene (ataxia-telangiectasia mutated gene) located on chromosome 11q22.58ATM is involved in detecting damage at DNA level and in cell cycle progression.59
Diagnostic criteria. The diagnosis of ataxia telangiectasia is based on compatible clinical symptoms and identification of the mutation in both alleles of the ATM gene,60 although as mentioned previously, the risk of PDAC does not require a biallelic mutation.61
Familial atypical multiple mole melanoma syndromeThis syndrome is associated with mutation of a tumour suppressor gene (p16/CDKN2A), and is characterised by the development of numerous dysplastic nevi and melanoma. Patients with this syndrome have a 13–22-fold increased risk of pancreatic cancer compared to the general population.62 A recent study in the Spanish population found a higher prevalence of pancreatic cancer (prevalence rate 2.97, p=0.006) in patients with multiple melanoma who were CDKN2A carriers compared to patients with multiple melanoma with no identified mutation.63
Genetics. Autosomal dominant inheritance with incomplete penetrance. Associated with germline mutations in the p16/CDKN2A gene located on chromosome 9p21. CDKN2A encodes 2 proteins: p16 and p14ARF. Protein p16 is a negative regulator of cell cycle progression.64
Diagnostic criteria. The incidence of mutations in CDKN2A is in fact higher in individuals with 3 or more melanomas and/or in families with at least 1 member with melanoma and another 2 or more FDR or SDR with a diagnosis of PDAC.
The diagnostic clinical criteria for FAMMM are:
- •
Malignant melanoma in >1 FDR and presence of >50 nevi and nevi with atypical histological characteristics.
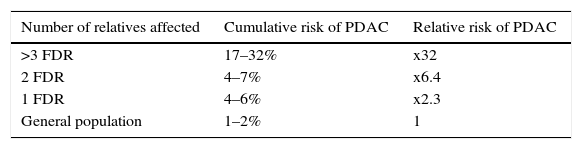
FPC is the most common syndrome of PDAC, in which familial aggregation of this neoplasm is observed with no identified genetic cause. The increased risk of developing this neoplasm is determined by the number of family members affected (Table 2).65
Genetics. The causal germline mutation is unknown.
Diagnostic criteria. The criteria that define FPC are:
>2 FDR regardless of age, or
>3 FDR or SDR or third degree relatives (TDR), regardless of age.
Screening for hereditary and/or familial pancreatic ductal adenocarcinomaIndividuals with a family history of PDAC are considered to be at higher risk for developing cancer; they should therefore be included in screening programmes under a multidisciplinary team. The aim of these programmes is to detect PDAC precursor lesions or early-stage cancer (intraductal papillary mucinous tumour [IPMT], intraepithelial pancreatic neoplasm grade 3 [PanIN3], T1N0M0).66
The IPMTs are potentially malignant intraductal epithelial neoplasms composed of mucin-producing columnar cells, with papillary proliferation, cyst formation and variable grades of cellular atypia; these lesions can affect both the main pancreatic duct and the secondary branches. Due to the risk of malignancy (70% in main duct IPMT), these lesions must be diagnosed and treated appropriately.67
Although there is no firm evidence that screening is associated with a reduction in PDAC-related mortality, patients who will be candidates for surgery if a suspicious lesion is found in the study should be included in screening programmes.
Screening is recommended in the following cases68:
- •
Members of families with HPC with known germline mutation:
- 1)
Individuals diagnosed with PJS or hereditary pancreatitis.
- 2)
Carriers of a mutation in p16/CDKN2A, BRCA1, BRCA2, ATM, PALB2, MSH2, MLH1, MSH6, PMS2 or APC, with a family history of PDAC.
- 1)
- •
Members of families with FPC: individuals with 2 or more FDR or individuals with 3 or more family members (FDR, SDR or TDR) with PDAC, with at least one of them being an FDR.
Endoscopic ultrasound and magnetic resonance imaging are currently the studies of choice for screening programmes, as these have shown the highest detection rates of PDAC precursor lesions. The first option is endoscopic ultrasound (given its ability to detect lesions measuring less than 10mm), with magnetic resonance imaging recommended as an alternative.66,69
Screening programmes generally commence at the age of 40, or at an age 10 years younger than the youngest family member affected. However, 2 recent studies66,70 have shown that the diagnostic yield is significantly higher after age 65 (35% vs 3%); increasing the age for the first examination to 50 years, therefore, seems justified, unless the family members affected have developed the cancer at an early age. Risk of early-onset cancer is higher in 2 situations, each with a different screening strategy: (a) screening for PDAC should be started at age 30 years in patients with PJS, (b) screening for PDAC should be started at age 40 years in patients with HP. Annual screening is recommended in all cases.
If a suspicious lesion is detected, there is no consensus between partial or total pancreatectomy. One alternative is partial resection with total pancreatic resection if intraoperative histopathology confirms PDAC, multifocal PanIN2/3 or IPMT with high grade dysplasia. The decision regarding the type of surgical intervention should be made on an individual basis, taking into consideration the presence of exocrine and endocrine pancreatic insufficiency. Prophylactic total pancreatectomy is not indicated in asymptomatic patients55,66 (Fig. 3).
Future outlook
The underlying genetic cause has not been identified in a high percentage of families with GC and PC aggregation. Due to the difficulty in establishing diagnostic criteria for these families, combined with the fact that the same phenotype can be caused by various genes and, conversely, a single gene can be responsible for various phenotypes, identification of these diseases in clinical practice is challenging.
A recent study,6 for example, found that patients who met clinical criteria for HDGC, with a negative CDH1 genetic study, presented mutations in other genes (CTNNA1, BRCA2, STK11, among others), suggesting that HDGC syndrome can be defined by mutations in CDH1 as well as in other related genes.
The current availability of next generation sequencing technology will likely lead to the discovery of new genetic mutations responsible for hereditary gastric and pancreatic cancer. Similarly, with commercial next generation sequencing panels soon to be available, the approach to genetic testing and the complexity of genetic counselling will change significantly. The use of multigene panels will allow the simultaneous analysis of various genes at a lower cost. One of the main challenges that this new technology will bring will be interpretation of the information and management of numerous variants of unknown significance, requiring greater multidisciplinary collaboration.
These new diagnostic tools will most likely improve the identification and characterisation of this subgroup of patients at high risk of GC and PC in the near future, giving greater insight into the pathogenesis and behaviour of this neoplasm, and enabling clinicians to offer the best management strategies available (screening, surveillance and treatment) in order to reduce cancer incidence and mortality.
ConclusionThe different syndromes that predispose an individual to hereditary gastric cancer and/or hereditary pancreatic cancer are rare diseases but with significant morbidity and mortality. Several of these are associated with known mutations, but a genetic cause cannot be identified in most. Early diagnostic and surveillance criteria designed to reduce the morbidity and mortality caused by these neoplasms can be established on the basis of the syndrome and/or family history. For these reasons, surveillance of family members at high risk of these diseases should be performed in specialised centres, and if possible, within a research protocol.
Conflict of interestsThe authors declare that they have no conflict of interests.
Please cite this article as: Leoz ML, Sánchez A, Carballal S, Ruano L, Ocaña T, Pellisé M, et al. Síndromes de predisposición a cáncer gástrico y cáncer pancreático. Gastroenterol Hepatol. 2016;39:481–493.
