




The diagnosis of suspected Lynch syndrome without known mutation, or Lynch-like syndrome (LLS), is established when there is loss of expression of DNA repair proteins (mismatch repair or MMR) or high microsatellite instability, not associated with MLH1 methylation or a BRAF mutation, but without evidence of mutation of the genes encoding these proteins in germline analysis. It is a heterogeneous condition which can range from true hereditary syndromes to cases of sporadic colorectal cancer (CRC). In such cases, multi-gene panels play an important role in differentiating the two.
We present the cases of two patients initially suspected of having LLS, but with a pathogenic mutation in other genes related to DNA repair after massive sequencing of these panels.
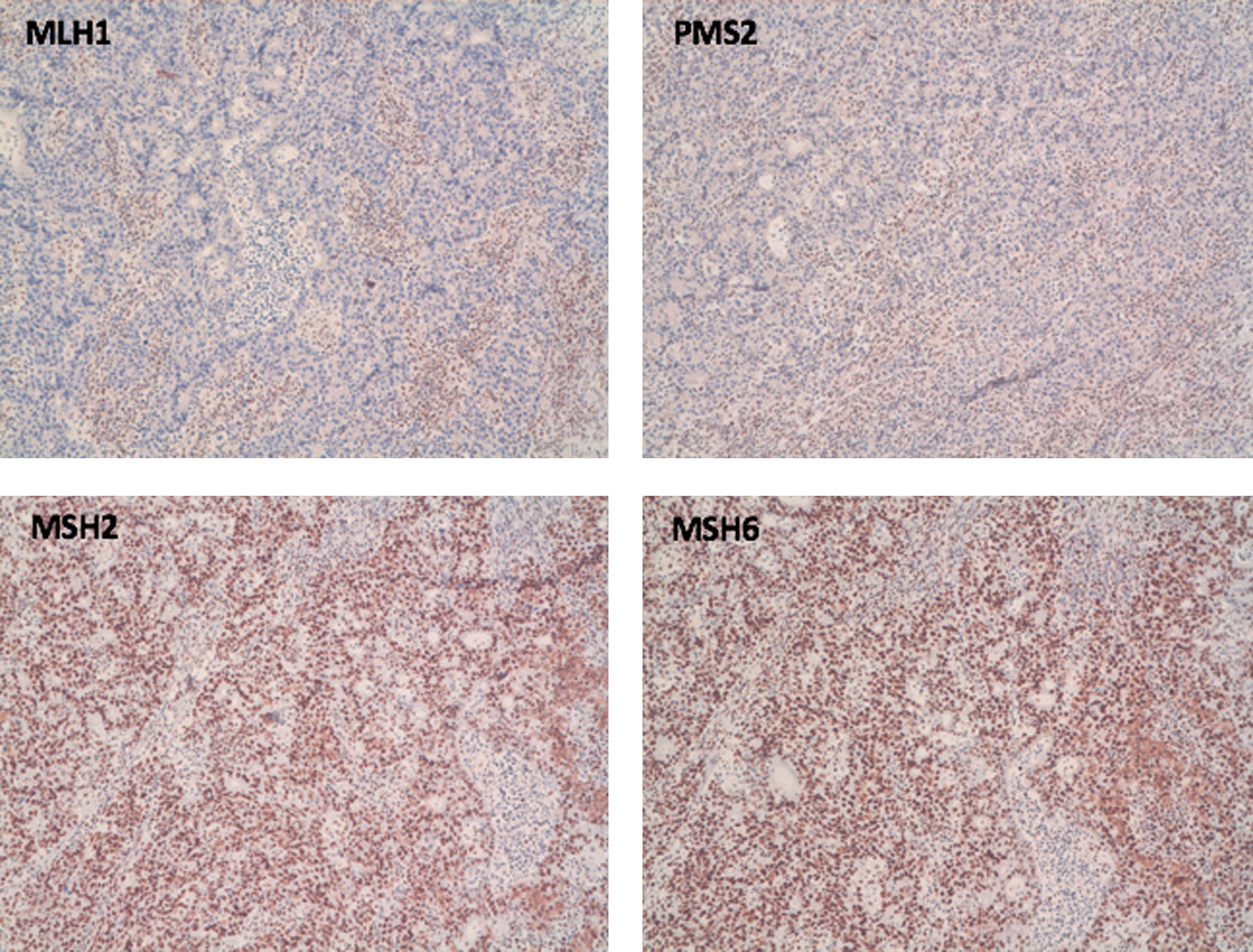
Case 1A 70-year-old female with a history of first- and second-degree relatives with endometrial cancer, breast cancer and gastric cancer, diagnosed with CRC with loss of nuclear expression of MLH1 and PMS2 (Fig. 1). A BRAF mutation study showed native BRAF, and so a genetic study was performed. Germline analysis with a multigene panel showed no mutations in the MLH1, MSH2, MSH6, PMS2 and EPCAM genes, but a heterozygous variant c.280–821del (p.Leu274Phefs*16) was detected in the MRE11A gene, and considered as possibly pathogenic.
Case 2 with intact nuclear expression for MSH2 and MSH6 (bottom photos).")
A 54-year-old female referred with a previous history of CRC at age 23 and a first-degree relative with CRC at age 62. As in the other case, loss of MLH1 expression was detected, but with germline analysis of the genes related to normal Lynch syndrome. However, a heterozygous variant c.1343T>G (p.Leu448Ter) was obtained in the BARD1 gene, described as a possible pathogenic variant.
DiscussionWith the recommendation of universal molecular screening for Lynch syndrome, it is increasingly common to find molecular alteration not accompanied by a germline mutation. There may be different causes for this situation: (1) atypical germline alterations not identified with the current means which cause a somatic mutation of the remaining alleles; (2) germline alterations in other genes (such as MUTYH, POLD1, POLE) which could affect the MMR system; or (3) somatic alterations in oncogenes, biallelic somatic mutations in MMR genes, or a combination of the two. The clinical behaviour of LLS is not fully understood. It is known, however, that the incidence of CRC in families with LLS is higher than in families with sporadic CRC, but lower than in families with Lynch syndrome. In a study of 160 patients with LLS, the demographic, clinical and histological characteristics were similar, regardless of family history.1
Within this heterogeneous disorder we distinguished two types of patient: those with a family history, which suggests a hereditary syndrome, but without evidence of a family mutation; and those with no significant family history of CRC in whom the only suspicious element in terms of Lynch syndrome is the molecular alterations. In these cases, the most common cause is usually a somatic double mutation in the MMR genes. For that reason, some authors propose assessing somatic mutations to classify them as sporadic or hereditary.2 The latest clinical practice guidelines for the diagnosis and prevention of CRC, in addition to the analysis of somatic mutations, propose using multi-gene panels to exclude germline mutations in other genes,3 as performed in the cases presented here.
The MRE11 gene plays an important role in the response to DNA damage and the repair of double-strand breaks. MRE11 deficiency can cause microsatellite instability through defective interaction with MLH1 and lead to its inactivation in MMR-deficient tumours.4 The interaction between BARD1 and BRCA1 promotes the tumour suppressor function by activating the repair of double-strand breaks and the initiation of apoptosis.5
Patients with LLS are therefore a heterogeneous group of patients in whom the study by massive sequencing can help to distinguish a true inherited syndrome from sporadic CRC.
Please cite this article as: Adán-Merino L, Valentín-Gómez F, Tirado-Zambrana S, Zaera-de la Fuente C, Crivillén-Anguita O, Aldeguer-Martínez B. Lynch-like syndrome: síndrome de Lynch ¿sin mutación conocida? Gastroenterol Hepatol. 2021;44:376–377.
