




Protein-losing enteropathy (PLE) is a syndrome characterised by protein leakage through the gastrointestinal tract that causes a decrease in plasma oncotic pressure and clinically manifests with oedema, ascites, pleural effusion and/or pericardial effusion.1
We present the case of a six-year-old male patient from a rural community, hospitalised for asthenia, adynamia, diarrhoea and vomiting for seven days. He had a history of consumption of unpasteurised milk and a complete vaccination schedule. A physical examination revealed moderate dehydration, abdominal distension and pain, oedema of the lower limbs, and decreased frequency of bowel sounds. Eutrophic nutritional status (weight for age 96% and height for age 109%). An abdominal X-ray revealed abundant colon gas and multiple air-fluid levels. Laboratory studies reported haemoglobin 16.7 g/dl (normal [n]: 11.5–13.5), haematocrit 46.4% (n: 34–40), serum albumin 1.2 g/dl (n: 3.5–5.2), serum calcium 6.5 mg/dl (n: 8.8–10.1), serum immunoglobulins IgG 177 mg/dl (n: 560–1,307), IgA 82 mg/dl (n: 26–232), IgE 37.9 Ul/ml (n: 0.3–17.6) and serum cholesterol 81 mg/dl (n: 108–187). Urinalysis showed no protein and liver function tests were normal. On the fifth day of hospitalisation, an exploratory laparotomy was performed due to clinical data revealing intestinal subocclusion, and multiple lymphadenopathies in the mesentery were identified by the procedure. Subsequently, he presented with bilateral pleural effusion and ascites. Bilateral thoracentesis was performed, and assisted ventilation, albumin infusion, and diuretics were applied. Pleural fluid showed transudate characteristics: leukocytes zero, glucose 95 mg/dl and lactic dehydrogenase 14 IU/ml.
With this information, the diagnosis of PLE was integrated, in the following weeks hypoalbuminaemia, hypogammaglobulinaemia and hypocalcaemia persisted and he presented with profound lymphopaenia (total serum lymphocytes 260 cells/μl [n: 1,500–5,900], CD3 T lymphocytes 155 cells/μl [n: 700–4,200] and CD19 B lymphocytes 49 cells/μl [n: 200–1,600]).
To diagnose the different causes of PLE, stool studies were requested (stool cultures: two; fresh stains: two, Kinyoun stain, culture in selective medium for Campylobacter sp., Clostridium difficile A/B toxin) with negative results. Serum polymerase chain reaction (PCR) for cytomegalovirus and serology for human immunodeficiency virus were negative. Negative anti-nuclear antibodies. Serum amylase, lipase and complement (C3 and C4) within normal parameters. Ultrasound without structural abnormalities. Gadolinium-enhanced magnetic resonance imaging revealed thickened loops in the distal jejunum and proximal ileum (4–6 mm in diameter) with oedema in the root of the mesentery and reactive appearing lymph nodes.
Upper gastrointestinal endoscopy revealed patchy white lesions (snowflake images) in the first and second portions of the duodenum and chylous reflux; the histopathological study reported active duodenitis with widening of the villi, chronic ileitis, and tortuous and dilated mucosal lymphatic vessels (Fig. 1); four eosinophils were observed per high-power field. The histopathological study of the mesenteric lymph node showed dilatation and sinus proliferation compatible with vascular transformation.
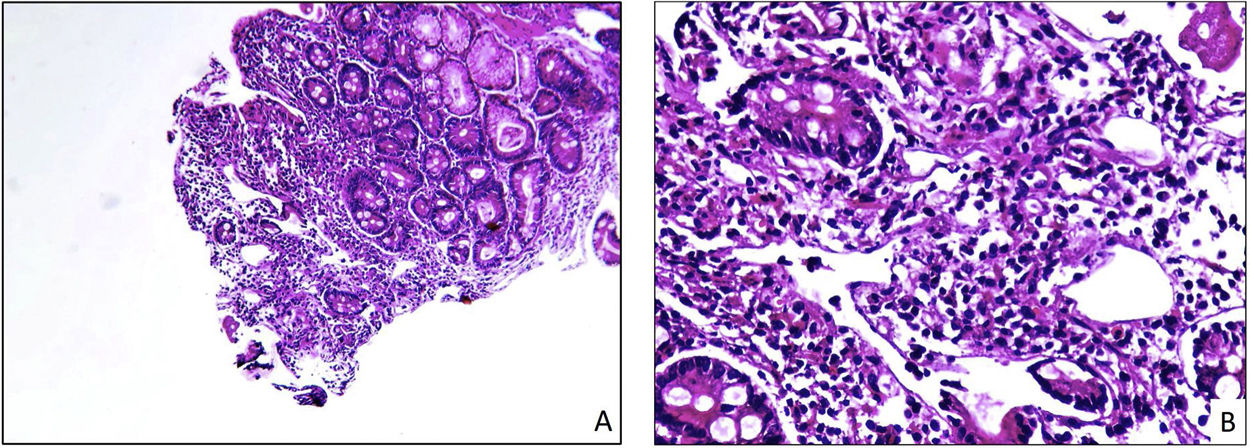
 Intermediate-magnification photomicrograph (×10) in which a fragment of duodenal mucosa with lymphangiectasia can be seen. Note the oedema and lymphatic dilatation seen in the central area of the image. (B) High-magnification photomicrograph (×40) in which a fragment of duodenal mucosa affected by lymphangiectasia can be seen. Note the lymphoplasmacytic inflammatory infiltrate of the lamina propria, as well as the presence of tortuous and dilated lymphatic vessels.")
Histopathological image of duodenal biopsies with lymphangiectasia. (A) Intermediate-magnification photomicrograph (×10) in which a fragment of duodenal mucosa with lymphangiectasia can be seen. Note the oedema and lymphatic dilatation seen in the central area of the image. (B) High-magnification photomicrograph (×40) in which a fragment of duodenal mucosa affected by lymphangiectasia can be seen. Note the lymphoplasmacytic inflammatory infiltrate of the lamina propria, as well as the presence of tortuous and dilated lymphatic vessels.
Genetic sequences of the Mycobacterium tuberculosis complex were identified by PCR on intestinal tissue.
PLE is a manifestation of intestinal or systemic diseases and is caused by injury to the intestinal mucosa (ulcerative, inflammatory and/or allergic diseases) or by abnormalities in the lymphatic system (lymphangiectasias) that may be congenital (Waldmann’s disease) or secondary to obstruction (sarcoidosis, lymphoma, or inflammatory bowel disease) or to elevated venous pressure (congestive heart failure or constrictive pericarditis). The diagnosis of PLE is established by the presence of signs and symptoms secondary to hypoalbuminaemia with no other apparent cause such as proteinuria, malnutrition, or liver failure. Other tools for diagnosis are the detection of alpha-1-antitrypsin in stool samples or functional imaging studies.1 The diagnosis of lymphangiectasia is made by histological examination of intestinal tissue.2
In patients with mesenteric lymphadenitis due to tuberculosis, the increase in intestinal lymphatic pressure causes dilatation of the lymphatic vessels and leakage of proteins to the surface epithelium.3 Although, in adult patients it has been described that M. tuberculosis can exacerbate the symptoms of primary lymphangiectasia2 or cause secondary lymphangiectasia3 this association has not been reported in children.
For the treatment of patients with PLE, it is recommended that an adequate nutritional status be maintained and the underlying cause be treated.1 In our patient, it was not possible to determine whether the tuberculous infection arose from the immunosuppression caused by the PLE or if it was the cause of intestinal lymphatic dilatation. The patient received treatment with antitubercular drugs, intravenous immunoglobulin, and a low-fat, high-protein diet with medium-chain triglycerides and fat-soluble vitamins. Seven weeks later, he had normal serum albumin, calcium, and cholesterol values; increased lymphocyte count and an absence of signs and symptoms.
Due to the presence of eosinophils in the intestinal biopsy, serum tests were performed for the detection of specific IgE antibodies for food allergens, without reactive results. Similar to other reports, the improvement of the patient with antitubercular treatment and without the administration of corticosteroids suggests a relationship between intestinal eosinophilia and tuberculous infection.
In Mexico, consumption of unpasteurised milk is a common practice,4 and this habit increases the risk of tuberculosis,5 mainly in children and in patients with immunodeficiencies. For the diagnosis of intestinal tuberculosis, clinical manifestations (fever, abdominal pain, hepatomegaly, and diarrhoea), exposure to a source of infection, and identification of the bacteria in intestinal biopsy or peritoneal fluid must be considered. A strongly suggestive histological finding is caseating granulomas, but these are observed in fewer than half of the cases. In paediatric patients with PLE and risk factors for tuberculosis, we suggest taking a diagnostic approach for this infection.
Please cite this article as: Lona-Reyes JC, Torres-Molina S, Flores-Fong LE, Estrada-Arce EV, Rivera-Chávez E, Núñez-Núñez ME, et al. Enteropatía pierde-proteínas en un paciente preescolar con tuberculosis intestinal. Gastroenterol Hepatol. 2022;45:204–205.
