









No studies evaluating the rapidity of response to biological therapies are available for Crohn's disease (CD). The aim of this study was to evaluate rapidity of onset of clinical response and impact on quality of life (QoL) of adalimumab therapy in adult anti-TNF-naïve patients with moderately-to-severely active CD.
Patients and methodsRAPIDA was an open-label, single-arm, prospective, multicenter clinical trial. Adult patients with moderately-to-severely active luminal CD, anti-TNF-naïve, and unresponsive to conventional therapy were treated with adalimumab. Clinical disease activity, QoL and inflammatory biomarkers were measured at day 4, and weeks 1, 2, 4, and 12 after treatment initiation.
ResultsEighty-six patients were included in the intention-to-treat (ITT) analyses. Clinical disease activity was reduced from a median of 9.0 points to 6.0 points at day 4. Clinical response (≥ 3-point reduction in the Harvey-Bradshaw Index, HBI) was achieved by 61.6% (d4) and 75.6% (w1) of patients in the ITT population (median 2.5 days) and with non-responder imputation (NRI), by 55.8% and 53.4%, respectively. The proportion of patients in clinical remission (HBI<5) at weeks 2 and 4 in the ITT population was 54.7% and 62.8%, respectively (median 7.0 days), and 38.4% and 45.3% in the NRI population. All QoL scores significantly improved and inflammatory biomarkers significantly decreased from day 4 onwards (p<0.0001).
ConclusionRapid clinical response and remission, improvement in QoL and fatigue, and a reduction of inflammatory biomarkers were achieved with adalimumab as early as day 4 in adult anti-TNF-naïve patients with moderately-to-severely active CD.
No hay estudios que evalúen la rapidez de la respuesta a las terapias biológicas en la enfermedad de Crohn (EC). El objetivo fue evaluar la rapidez del inicio de la respuesta clínica y el impacto en la calidad de vida (CdV) de la terapia con adalimumab en pacientes adultos con EC moderada-grave.
Pacientes y métodosRAPIDA fue un ensayo clínico abierto, de un solo brazo, prospectivo y multicéntrico. Se trató con adalimumab a pacientes adultos con EC luminal activa moderada-grave, sin tratamiento previo con anti-TNF y sin respuesta a terapia convencional. Se midieron la actividad clínica de la enfermedad, la CdV y los biomarcadores inflamatorios el día 4 y las semanas 1, 2, 4 y 12 tras el tratamiento.
ResultadosSe incluyeron 86 pacientes en los análisis por intención de tratar (IdT). La actividad clínica de la enfermedad se redujo de una mediana de 9,0 a 6,0 puntos en el día 4. La respuesta clínica (reducción ≥3 puntos en el Índice Harvey-Bradshaw, IHB) se dio en un 61,4% (d4) y un 75,6% (s1) de los pacientes IdT y en un 55,8% y un 53,4% con imputación de no respondedores (INR). La proporción de pacientes IdT en remisión clínica (IHB<5) en las s2 y s4 fue de 54,7% y 62,8%, respectivamente, y 38,4% y 45,3% en la población INR. Todas las puntuaciones de CdV mejoraron significativamente y los biomarcadores inflamatorios disminuyeron significativamente desde el día 4 (p<0,0001).
ConclusionesSe logró una rápida respuesta clínica y remisión, mejoría en la CdV y fatiga, y una reducción de los biomarcadores inflamatorios en los pacientes tratados con adalimumab ya en el día 4.
Crohn's disease (CD) is a chronic inflammatory bowel disease (IBD) characterized by chronic transmural inflammation. Symptoms are heterogeneous, and progression may lead to penetration, strictures and refractory disease, requiring repeated hospital admissions and surgical intervention.1,2
Clinical response and remission2,3 as well as the prevention of structural bowel damage4 have long been considered targets for CD therapy. Although much attention is currently being paid to other targets, such as mucosal healing,5 perhaps they are not as adequate as the clinical response to assess the rapidity of the treatment effect.
Speed of response to treatments for IBD is now considered a major field of interest, due to the importance of achieving symptom reduction quickly, in order for patients to have a rapid return to normal life, and to prevent potential life-threatening complications in patients with severe disease. Patients with IBD live in a constant state of anxiety, and knowing that they will experience rapid relief from disease symptoms is important to them. In fact, it has been shown that clinical response is associated with improved quality of life (QoL),6 as well as a reduction in fatigue and depression,6–8 the benefits of which may be associated with lower disease-related costs.7 Time to therapeutic response is an important yet underestimated factor in the day-to-day management of IBD.9 However, no previous studies specifically designed to evaluate the rapidity of response to adalimumab or any other biological therapy in patients with active CD have been conducted, and treatment response data earlier than week 1 are lacking.9
With a vast array of different therapies, approved or in development, it is important to identify the pros and cons of the available drugs to ensure that patients receive the most appropriate treatment. Important advances have been made in the understanding of the pathogenesis of IBD, leading to the introduction of new biological drugs in therapeutic algorithms.2,3,10 Adalimumab is a fully human anti-TNF monoclonal antibody (mAb) that has proven to be effective in inducing and maintaining clinical response and remission in patients with moderate to severe CD.11 The results of the CLASSIC 1 trial12 suggested a rapid response to adalimumab in CD patients, with clinical response rates (reduction in CD Activity Index [CDAI]≥70 points) ranging from 32% to 40% across the adalimumab treatment arms at week 1.
The aim of this study was to evaluate the rapidity of onset of clinical response to standard adalimumab therapy. Eligible patients were adult CD patients with moderate-to-severe activity (≥8 points in the Harvey-Bradshaw Index [HBI]), with no response to complete and adequate therapy with corticosteroids and/or immunosuppressants. Considering the impact of disease activity on the QoL—the most preferred outcome for patients13—and fatigue,7,8 these aspects were also specifically investigated. Secondary endpoints also included assessment of the early effects of adalimumab on the levels of C-reactive protein (CRP) and fecal calprotectin (FC), markers of disease-related inflammation, the normalization of which has been proposed as an adjunctive target of CD therapy.5,14,15
Patients and methodsStudy design and ethical considerationsRAPIDA was an open-label, single-arm, prospective, phase 4 clinical trial conducted in 21 participating centers throughout Spain, from May 27, 2014, to January 23, 2017. The study enrolled patients with active luminal CD who were unresponsive to a previous adequate course of therapy with corticosteroids and/or immunosuppressants. The Institutional Review Board of Hospital Josep Trueta (Girona, Spain) approved the study protocol and the study-specific informed consent, which was signed and dated by each participating subject before any study procedures were performed or any medications were withheld from the subject in order to participate. The study was conducted in accordance with the applicable regulations and guidelines, and the principles of the Declaration of Helsinki. ClinicalTrials.gov identifier: NCT02148718.
PatientsPatients (n=100) were aged from 18 to 75 years and presented with moderately-to-severely active luminal CD (HBI≥8)16 diagnosed≥4 months, with documented clinical symptoms and endoscopic/radiological findings according to standard criteria, and unresponsive to a previous complete and adequate course of therapy with a corticosteroid and/or an immunosuppressant. Selection criteria were based on the approved indication,17 previous clinical trials,12 and guidelines from the European Medicines Agency (EMA). A full description of inclusion and exclusion criteria is available in the supplementary material.
Induction and maintenance treatmentThe doses of adalimumab (HUMIRA®, AbbVie Inc. North Chicago, Illinois, U.S.A.) used in the study were selected according to the product prescribing information for CD.17 All subjects received an open-label adalimumab induction dose of 160mg at week 0 (baseline), 80mg at week 2, and 40mg every other week from week 4 through week 10 (inclusive). Subjects did not receive the study drug at their last visit (week 12/premature discontinuation). After induction treatment, intensification to 40mg weekly was allowed at the investigator's discretion (Fig. 1A).
 Study design. The treatment schedule and questionnaires and scores administered at each visit are indicated. BL, baseline; D, day; D-FIS, Fatigue Impact Scale for Daily Use; EQ-5D, EuroQol-5 Dimensions questionnaire; EOW, every other week; HBI, Harvey–Bradshaw Index; IBDQ-36, 36-item Inflammatory Bowel Disease Questionnaire; W, week. (B) Flow diagram of patient disposition. ITT, intention-to-treat; PP, per-protocol.")
(A) Study design. The treatment schedule and questionnaires and scores administered at each visit are indicated. BL, baseline; D, day; D-FIS, Fatigue Impact Scale for Daily Use; EQ-5D, EuroQol-5 Dimensions questionnaire; EOW, every other week; HBI, Harvey–Bradshaw Index; IBDQ-36, 36-item Inflammatory Bowel Disease Questionnaire; W, week. (B) Flow diagram of patient disposition. ITT, intention-to-treat; PP, per-protocol.
Concomitant treatments for CD were allowed if used at a stable dose (see supplementary material for a full description of the concomitant treatments that were permitted or prohibited). If the patient received oral budesonide (maximum dose of 9mg/day) or oral prednisone (or equivalent, maximum dose of 40mg/day), it was required that the dose has not increased in the last two weeks (dose reduction was allowed). If additional steroids were needed (as well as other rescue therapies such as cyclosporin, infliximab or surgery), a patient will be considered as treatment failure, but followed until final visit. Enteral nutrition via tube feeding as the entire nutritional intake prior to screening or baseline was not allowed.
Study procedures and variablesPatient visits were scheduled at baseline and on day 1, day 4, week 1, week 2, week 4, and week 12 or at premature discontinuation. Safety follow-up was performed 70 days after study completion or early discontinuation (Fig. 1B).
Complete medical and surgical histories of patients were collected at baseline, including tobacco and alcohol consumption, and detailed history of exposure to Mycobacterium tuberculosis. Chest X-ray was performed at the screening visit. Demographic details (age, sex, race) and electrocardiograms were obtained at screening/baseline. At each visit, anthropometric data were collected and a full physical examination was performed. The investigator completed the HBI (directly correlated with the CDAI)16,18 to assess disease activity, which was scored as follows: remission, <5; mild disease, 5–7; moderate disease, 8–16; and severe disease, >16. Laboratory tests were centrally performed; samples were obtained at each visit, with particular attention to FC, erythrocyte sedimentation rate (ESR), and CRP.
To assess changes in QoL and fatigue, 3 self-administered questionnaires were completed by the patient at all scheduled visits, except on day 1 (Fig. 1A): (i) the EuroQol-5 Dimensions questionnaire (EQ-5D; composed of two tools, the EQ-5D descriptive system [0 indicating “dead” and 1 indicating “full health”] and the EQ visual analog scale [EQ VAS, 0 indicating “the worst health state” and 100 indicating “the best health state”)19; (ii) the 36-item Inflammatory Bowel Disease Questionnaire (IBDQ-36; ranging from 36 to 252 points, with the highest score indicating the best QoL in relation to symptoms)20; and (iii) the Fatigue Impact Scale for Daily Use (D-FIS; scored from 0 to 32, with the highest score indicating the highest level of fatigue).21
EndpointsThe primary endpoint was the proportion of patients with a clinical response at day 4, defined as a decrease of ≥3 points on the HBI. Secondary analyses included: (1) proportion of patients with a clinical response at week 1; (2) proportion of patients with clinical remission (HBI <5) at weeks 2 and 4; (3) changes in QoL measurements (EQ-5D and IBDQ-36) from baseline through week 12; (4) changes in fatigue measurements (D-FIS) from baseline through week 12; (5) changes in laboratory biomarkers of inflammation from baseline through week 12, including complete blood count, ESR, CRP, FC, and coagulation parameters (including fibrinogen); (6) correlation between clinical response at day 4 and week 1 with remission at week 12; and (7) safety variables, which included adverse events (AEs; severity defined in the supplementary material) throughout the study until 70 days after the last treatment dose.
Statistical proceduresSample size was calculated assuming an expected response of 40% at day 4, based on a previous study,12 a standard error of 5% (95% confidence interval [CI], 30–50%), and a 5% dropout rate. With these parameters, 98 patients were needed.
The intention-to-treat (ITT) population (patients who received ≥1 dose of study treatment) was the primary population for efficacy and QoL analyses. Multiple imputation (modified ITT population, mITT) and non-responder imputation (NRI population) techniques were used for handling missing data. A per-protocol (PP) population, consisting of patients included in the ITT population who did not have major protocol deviations, was also analyzed. The safety population included all patients who received ≥1 dose of adalimumab.
Descriptive statistics were performed and quantitative data were summarized as size, mean, standard deviation (SD), 2-sided 95% CI, median, interquartile range (IQR), minimum, and maximum. Qualitative variables were summarized as absolute and relative frequencies in the entire population. The Fisher's exact test (2-sided) or chi-square test was used as appropriate to explore the associations of categorical data. The Student t test or Wilcoxon signed-rank test (for paired data) was used as applicable to explore the associations of numerical data in patients at week 0 versus week 12. The Shapiro–Wilks test was used to determine if the continuous measure followed a normal distribution. Kaplan–Meier analysis was performed to determine the time to event and the McNemar test was performed to evaluate the association between a rapid response at day 4 and week 1 with remission at week 12. Age, weight and body mass index (BMI), immunosuppressant use at baseline, intestinal area involved at baseline, and CD duration were used as covariates in univariate regression models in order to adjust the time to clinical response as assessed by HBI. The significance level was established at a value of α=0.05. All analyses were undertaken using SAS for Windows version 9.4 (SAS Institute, Cary, NC).
ResultsOne hundred patients from 21 sites were initially enrolled, of which 14 were screening failures, none of whom received study treatment. Eighty-six patients continued in the trial and received at least one dose of the study treatments, constituting the intention-to-treat (ITT) and safety populations. Of them, 12 patients discontinued the study prematurely and 19 were considered protocol deviations (16 received prohibited concomitant medication and 3 did not meet the study inclusion/exclusion criteria). Therefore, 55 patients constituted the per protocol (PP) population. The flow diagram of patient disposition is shown in Fig. 1B.
At baseline, the mean time from diagnosis was 5.6±6.3 years, and the mean patient age was 37.9±12.2 years. CD lesions were mainly located in the ileum (34.4%) or in the ileum and colon (40.7%). Thirty-seven patients (43.0%) had extraintestinal manifestations, most of whom had only one (n=29; 78.4%). All patients had moderate disease at initiation (HBI index), and had moderate scores for QoL (EQ-5D and IBDQ-36 questionnaires). Baseline characteristics are summarized in Table 1 (additional data in the supplementary material).
Baseline clinical and demographic characteristics and QoL scores of patients.
| Characteristic | n=86 |
|---|---|
| Female – n (%) | 49 (57.0%) |
| Caucasian – n (%) | 83 (96.5%) |
| Age, years – mean±SD | 37.9±12.2 |
| Never smoker – n (%) | 34 (39.5%) |
| Ex-smoker – n (%) | 20 (23.3%) |
| Years smoking (cigarettes/day) – mean±SD | 14.00±7.63 (11.06±7.37) |
| Months without smoking – mean±SD | 65.26±94.49 |
| Current smoker – n (%) | 31 (36.0%) |
| Years smoking (cigarettes/day) – mean±SD | 18.78±10.97 (9.86±5.22) |
| Body mass index, kg/m2– mean±SD | 24.6±4.4 |
| Time from diagnosis, years – mean±SD | 5.6±6.25 |
| Age at diagnosis – n (%)a | |
| A1 (<16 years) | 2 (2.3%) |
| A2 (17–40 years) | 69 (80.2%) |
| A3 (>40 years) | 14 (16.3%) |
| Missing – n (%) | 1 (1.2%) |
| Behavior at diagnosis – n (%) | |
| B1 – Non-stricturing, non-penetrating CD | 75 (87.2%) |
| B2 – Stricturing disease | 7 (8.1%) |
| B3 – Penetrating disease | 3 (3.5%) |
| Missing | 1 (1.2%) |
| Location at diagnosis – n (%) | |
| L1 – Ileal | 30 (34.4%) |
| L2 – Colonic | 18 (20.9%) |
| L3 – Ileocolonic | 35 (40.7%) |
| L4 – Isolated upper disease | 2 (2.3%) |
| Missing | 1 (1.2%) |
| Presence of extraintestinal manifestations – n (%) | 37 (43.0%) |
| Harvey–Bradshaw Index score – median (IQR) | 9.00 (8.0–10.0) |
| FC, μg/g – median (IQR) | 795 (385–1633) |
| CRP, mg/L – median (IQR) | 4.87 (2.21–10.85) |
| Concomitant medication at baseline – n (%) | 74 (86.1%) |
| Immunosuppressants | 31 (36.0%) |
| Immunosuppressants and corticosteroids | 29 (33.7%) |
| Other concomitant medication | 14 (16.3%) |
| EQ-5D index score – median (IQR) | 0.68 (0.47–0.74) |
| EQ-5D visual analog scale – median (IQR) | 55.0 (40.0–70.0) |
| IBDQ-36 overall score – median (IQR) | 142.5 (118.0–170.0) |
| D-FIS score – median (IQR) | 15.0 (7.0–21.0) |
According to Montreal classification at diagnosis. Data provided as n (%) unless otherwise stated. CD, Crohn's disease; CRP, C-reactive protein; D-FIS, Fatigue Impact Scale for Daily Use; EQ-5D, EuroQol-5 Dimensions questionnaire; FC, fecal calprotectin; IBDQ-36, 36-item Inflammatory Bowel Disease Questionnaire; IQR, interquartile range (Q1–Q3); SD, standard deviation.
Clinical response was achieved by 61.6% and 75.6% of patients in the ITT population at day 4 and week 1, respectively. In the mITT population, the percentages of patients who achieved clinical response at the same time points were 61.2% and 58.8%, respectively, while in the NRI population, in which response was imputed as non-responder in 15 patients, response was achieved by 55.8% and 53.4% of patients, respectively. In the PP population, clinical response at day 4 and week 1 was achieved by 69.1% and 83.6% of patients, respectively (Fig. 2A). The median time to clinical response was 2.5 days (95% CI, 1.0–7.0) in the ITT population (Fig. 2B) and 1.0 days in the PP population (95% CI, 1.0–4.0). The proportion of patients in the ITT population who were in clinical remission at weeks 2, 4 and 12 was 54.7%, 62.8% and 55.8%, respectively. In the mITT population, clinical remission was achieved by 45.9% of patients at week 2 and 36.5% at week 4. The percentages of clinical remission at the same time points in the NRI population were 38.4% and 45.3%, respectively (Fig. 2C). The median time to clinical remission in the ITT population was 7.0 days (95% CI, 4.0–13.0; Fig. 2D). The median time to clinical remission in the ITT population was 7.0 days (95% CI, 4.0–13.0; Fig. 2D); in the PP population, the median time to remission was 6.0 days (95% CI, 4.0–28.0). Achievement of response at day 4 or week 1 was significantly associated with clinical remission at week 12 in both the ITT and PP populations (ITT, p=0.011 and PP, p=0.008; McNemar test). It is worth noting that those patients who showed a clinical response at day 4, presented a 2.75 higher probability (95% CI, 0.897–8.435) of showing a clinical response at week 12. Moreover, 81.0% of all patients who showed a clinical response at day 4 or 14, also showed a clinical response at week 12.
 Percentage of patients with a clinical response at day 4 and week 1 in each study population. (B) Analysis of time to clinical response in the ITT population. (C) Percentage of patients with clinical remission at weeks 2, 4, and 12 in each study population. (D) Analysis of time to clinical remission in the ITT population. ITT, intention-to-treat; mITT, modified ITT; NRI, non-responder imputation; PP, per-protocol.")
Results of early clinical response and clinical remission. (A) Percentage of patients with a clinical response at day 4 and week 1 in each study population. (B) Analysis of time to clinical response in the ITT population. (C) Percentage of patients with clinical remission at weeks 2, 4, and 12 in each study population. (D) Analysis of time to clinical remission in the ITT population. ITT, intention-to-treat; mITT, modified ITT; NRI, non-responder imputation; PP, per-protocol.
Five (5.81%) patients required intensification to adalimumab 40mg weekly within the treatment period; one patient from week 8 to week 10, 2 patients from week 6 to week 10, and 2 patients from week 4 to week 10. Dose intensification, which was done proactively based on clinical parameters, had no effect on the proportion of patients with clinical response at day 4 or week 1, or the proportion of patients with clinical remission at weeks 2 and 4.
HBI scores for disease activity improved significantly versus baseline scores at day 4 and weeks 1, 2, 4, and 12. The overall HBI score was reduced from a median of 9.0 points (IQR, 8.0–10.0) at baseline to 6.0 (3.0–8.0) at day 4 in the ITT population, and from 9.0 (6.0–17.0) to 5.0 (0.0–15.0) in the PP population (Fig. 3A). Similarly, median CRP and FC levels were significantly reduced from day 4 compared with baseline levels; reductions were maintained throughout the treatment period to week 12 (p<0.0001 at all time points; Fig. 3B and C). Additional details on complementary inflammation markers have been summarized in the supplementary material.
![Change in disease activity according to HBI and markers of inflammatory activity. (A) HBI (the lines on each outer end correspond to quartile 1 [Q1, 25% percentile] and quartile 3 [Q2, 75% percentile]), respectively. (B) CRP. (C) FC. The p values in these figures denote the difference between scores at each time point and the baseline score (Student](https://static.elsevier.es/multimedia/24443824/0000004500000003/v1_202204220605/S2444382422000384/v1_202204220605/en/main.assets/gr3.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNcRcp7AE8pxI7/lU7i5mKGl6o67DY/uL2+/IxZemV5MIjNMk1b+6FlN1Tfx8us3qJwTB8LGJbs5DFtHdfb4+Diehmnk/W8/Cg3JGh+yTENSt2iyS65GsQzBs2jlUn0SwWfPm1ZRc5rkBdrsu+pfi49XapFo+xonSUUmQAcBA5L4Z++A5SmRpeOPsdPacFH3dxUh/5OQVeEOfQ+Rvphkz7x6bj8S7av0YVZHGXTbTDoX9/pibLEjoZFA1oNmqKCHgmI= "Change in disease activity according to HBI and markers of inflammatory activity. (A) HBI (the lines on each outer end correspond to quartile 1 [Q1, 25% percentile] and quartile 3 [Q2, 75% percentile]), respectively. (B) CRP. (C) FC. The p values in these figures denote the difference between scores at each time point and the baseline score (Student")
Change in disease activity according to HBI and markers of inflammatory activity. (A) HBI (the lines on each outer end correspond to quartile 1 [Q1, 25% percentile] and quartile 3 [Q2, 75% percentile]), respectively. (B) CRP. (C) FC. The p values in these figures denote the difference between scores at each time point and the baseline score (Student's t test in A; signed-rank test in B and C). BL, baseline; D, day; CRP, C-reactive protein; FC, fecal calprotectin; HBI, Harvey–Bradshaw Index; W, week.
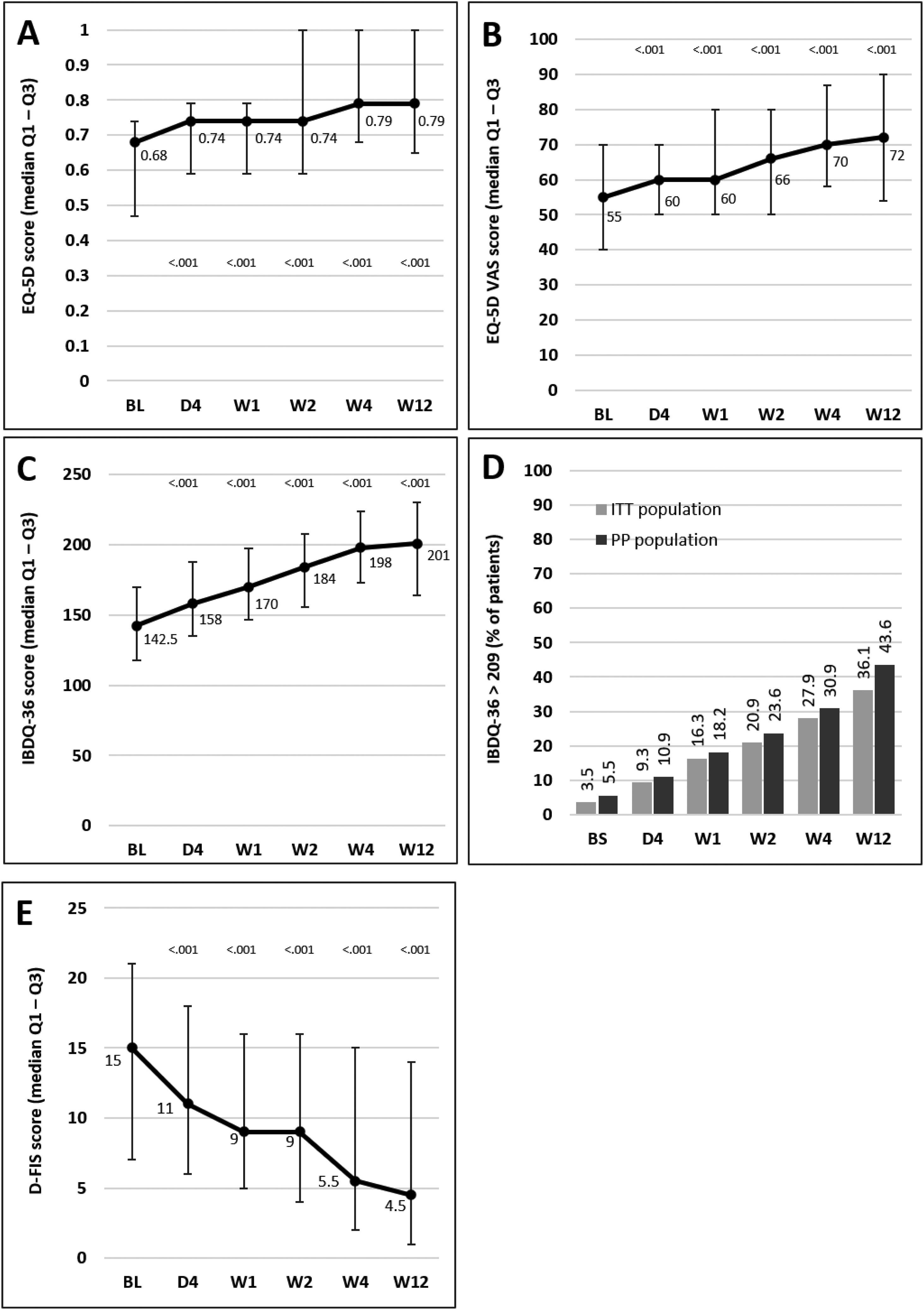
Median scores of the EQ-5D index and VAS and IBDQ-36 increased significantly versus baseline at all time points from day 4, indicating a progressive improvement in patient QoL. Fig. 4A–C illustrates the changes observed from baseline scores. Previous studies have suggested that IBDQ-36 scores >209 points correlate with a QoL comparable to that perceived by the general population.20 This threshold was achieved or exceeded by an increasing number of patients until the end of the study (Fig. 4D), and differences were statistically significant (p<0.05) from baseline scores at all time points in both the ITT and the PP populations (except for 10.9% in the PP population at day 4, which did not reach statistical significance). As measured by D-FIS, fatigue was progressively reduced (Fig. 4E), with significant differences in median scores at all time points compared with baseline. Scores of these questionnaires at each time point and by domain, where applicable, are provided in the supplementary material.
 209): significant differences were found at all time points in both populations compared with baseline (p<0.05), except for day 4 in the PP population. (E) Change in the D-FIS in the ITT population. The lines on each outer end correspond to quartile 1 (Q1, 25% percentile) and quartile 3 (Q3, 75% percentile), respectively. BL, baseline; D, day; D-FIS, Fatigue Impact Scale for Daily Use; EQ-5D, EuroQol-5 Dimensions questionnaire; IBDQ-36, 36-item Inflammatory Bowel Disease Questionnaire; QoL, quality of life; VAS, visual analog scale; W, week.' title='Change in QoL questionnaire and disease activity scores during the treatment period. (A) Change in the EQ-5D index score. (B) Change in the VAS of the EQ-5D. (C) Change in the IBDQ-36 score in the ITT population; p values in these figures denote the difference between scores at each time point and the baseline score (Student t test). (D) Restoration of the patient's health (defined as achievement of IBDQ-36 score>209): significant differences were found at all time points in both populations compared with baseline (p<0.05), except for day 4 in the PP population. (E) Change in the D-FIS in the ITT population. The lines on each outer end correspond to quartile 1 (Q1, 25% percentile) and quartile 3 (Q3, 75% percentile), respectively. BL, baseline; D, day; D-FIS, Fatigue Impact Scale for Daily Use; EQ-5D, EuroQol-5 Dimensions questionnaire; IBDQ-36, 36-item Inflammatory Bowel Disease Questionnaire; QoL, quality of life; VAS, visual analog scale; W, week.'/>
209): significant differences were found at all time points in both populations compared with baseline (p<0.05), except for day 4 in the PP population. (E) Change in the D-FIS in the ITT population. The lines on each outer end correspond to quartile 1 (Q1, 25% percentile) and quartile 3 (Q3, 75% percentile), respectively. BL, baseline; D, day; D-FIS, Fatigue Impact Scale for Daily Use; EQ-5D, EuroQol-5 Dimensions questionnaire; IBDQ-36, 36-item Inflammatory Bowel Disease Questionnaire; QoL, quality of life; VAS, visual analog scale; W, week.' title='Change in QoL questionnaire and disease activity scores during the treatment period. (A) Change in the EQ-5D index score. (B) Change in the VAS of the EQ-5D. (C) Change in the IBDQ-36 score in the ITT population; p values in these figures denote the difference between scores at each time point and the baseline score (Student t test). (D) Restoration of the patient's health (defined as achievement of IBDQ-36 score>209): significant differences were found at all time points in both populations compared with baseline (p<0.05), except for day 4 in the PP population. (E) Change in the D-FIS in the ITT population. The lines on each outer end correspond to quartile 1 (Q1, 25% percentile) and quartile 3 (Q3, 75% percentile), respectively. BL, baseline; D, day; D-FIS, Fatigue Impact Scale for Daily Use; EQ-5D, EuroQol-5 Dimensions questionnaire; IBDQ-36, 36-item Inflammatory Bowel Disease Questionnaire; QoL, quality of life; VAS, visual analog scale; W, week.'/>Change in QoL questionnaire and disease activity scores during the treatment period. (A) Change in the EQ-5D index score. (B) Change in the VAS of the EQ-5D. (C) Change in the IBDQ-36 score in the ITT population; p values in these figures denote the difference between scores at each time point and the baseline score (Student t test). (D) Restoration of the patient's health (defined as achievement of IBDQ-36 score>209): significant differences were found at all time points in both populations compared with baseline (p<0.05), except for day 4 in the PP population. (E) Change in the D-FIS in the ITT population. The lines on each outer end correspond to quartile 1 (Q1, 25% percentile) and quartile 3 (Q3, 75% percentile), respectively. BL, baseline; D, day; D-FIS, Fatigue Impact Scale for Daily Use; EQ-5D, EuroQol-5 Dimensions questionnaire; IBDQ-36, 36-item Inflammatory Bowel Disease Questionnaire; QoL, quality of life; VAS, visual analog scale; W, week.
Additional analyses were performed to determine the influence of various baseline factors on time to clinical response. HBI scores at baseline were significantly associated with time to clinical response in both the ITT and the PP populations (r=0.5953, p<0.0001 and r=0.4153, p=0.0014, respectively). However, the intestinal segment involved at baseline and disease duration was not correlated (p>0.05), and neither were the ages of patients or the BMI or weight at baseline. For the ITT population, the use of immunosuppressants as concomitant medication at baseline was not associated with the time to clinical response, that was comparable, 2.5 days, between those who were taking and those who were not taking concomitant immunosuppressants (p=0.2103; log-rank test). In this population, patients who were taking corticosteroids as concomitant medication at baseline showed, compared to those who were not taking concomitant corticosteroids, a slower clinical response, 4 days vs. 1 day; however, the difference was not statistically significant (p=0.2061; log-rank test).
Safety analysisDuring the study, 42.5% of patients presented with AEs, and a total of 87 AEs were reported, all of which were all mild or moderate in severity, except for 1 (worsening of CD). A full list of AEs is available in the supplementary material. Three serious AEs (SAEs) unrelated to the study drug, were reported: CD (severe), pyrexia (moderate), and pneumonia (mild). Eleven patients (12.8%) experienced 16 AEs that were assessed as being related to the study drug (Table 2). Four AEs led to temporary or permanent interruption of adalimumab: subocclusive syndrome (mild), hypersensitivity to the medication (moderate), cellulitis (moderate), and influenza (mild).
Adverse events related to the study drug.
| SOC term | Preferred term | Mild | Moderate |
|---|---|---|---|
| n (%) | n (%) | ||
| Eye disorders | Eyelid disorder | 1 (1.16%) | – |
| Immune system disorders | Drug hypersensitivity | – | 1 (1.16%) |
| Infections and infestations | Cystitis | 1 (1.16%) | – |
| Pharyngitis | 1 (1.16%) | – | |
| Rash pustular | 1 (1.16%) | – | |
| Respiratory tract infection | 1 (1.16%) | – | |
| Rotavirus infection | 1 (1.16%) | – | |
| Urinary tract infection | – | 1 (1.16%) | |
| Investigations | Weight decreased | 1 (1.16%) | – |
| Musculoskeletal and connective tissue disorders | Muscular weakness | 1 (1.16%) | – |
| Skin and subcutaneous tissue disorders | Alopecia | 2 (2.33%) | – |
| Eczema | 1 (1.16%) | – | |
| Psoriasis | – | 2 (2.33%) | |
| Skin lesion | 1 (1.16%) | – |
SOC, system organ class.
To our knowledge, this is the first study specifically designed to evaluate the rapidity of clinical response to a biologic therapy used in IBD at such an early time point. It therefore represents a new paradigm in the evaluation of therapies for this disease, in which rapidity of response may be of great relevance. In this study, we found that rapid clinical response was achieved as early as day 4. Improvement was assessed using HBI and confirmed by the rapid reduction in inflammatory markers of disease (CRP and FC) from day 4 and throughout the 12-week treatment period. Patients reported significant improvements in validated QoL instruments from day 4 of therapy, with scores that were sustained, or continued to improve, until the end of treatment. This is the first trial demonstrating such a rapid response to adalimumab in patients with luminal CD, and complements previous results that support the use of this mAb for the treatment of moderate to severe disease in patients who fail to respond to conventional therapy, in whom benefits in the medium- and long-term have already been reported.12,22,23
Such a quick response to adalimumab is promising for patients with CD, who need rapid normalization of their daily life and QoL. Additionally, reduced (optimal) time to therapeutic response may benefit the doctor–patient relationship, as well as patient adherence to treatment.9,24
The markers of disease activity used in this trial are based on the therapeutic targets in IBD recommended by the STRIDE group,5 such as the clinical and patient-reported (PRO) outcomes for CD, in which resolution of abdominal pain and diarrhea/altered bowel habits was considered key. The HBI index, well correlated with the CDAI index,18 was selected as an easy-to-use tool that takes into account these main symptoms (items 2 and 3; see supplementary material). HBI was complemented with 3 QoL PRO measures and surrogate laboratory biomarkers (CRP and FC levels), which are considered as adjunctive targets and useful markers for response to treatment.5 All these analytical endpoints have good correlation with ileocolonoscopy and imaging methods to evaluate inflammation, thus allowing assessment of rapid response.2
Patients achieved a median reduction in the HBI index of 3 points by day 4 (remission/mild disease), although further improvement at later time points was also observed. The median time to clinical response was as early as 2.5 days, and 50% of the ITT population was in remission within the first week of treatment, suggesting the rapidity of response to standard adalimumab. The effect on clinical parameters was paralleled by a quick and significant reduction of the inflammatory biomarkers CRP and FC at day 4, which was maintained throughout the 12 weeks of the study. FC has been proposed as a non-invasive marker for the direct evaluation of intestinal inflammation in patients with IBD,14,15,25 and has proven useful in the diagnosis of active inflammation in CD.2 On the other hand, a decrease in CRP levels is indicative of treatment response,14,26 and a good correlation has been shown between levels of this inflammatory marker and clinical and endoscopic disease activity in CD.2,27
An important finding in this study is the significant improvement in QoL and fatigue in patients as early as day 4 of adalimumab treatment, with an increase in global IBDQ-36 scores from a median baseline of 143–158. These results are in line with those of the CLASSIC-I study, in which patients treated with the same induction treatment increased their IBDQ-32 scores from a median baseline of 127 to >145 at week 1.12 A similar improvement was reported in the ACCENT-I trial with infliximab after 2 weeks of treatment.28 In fact, in the present study, QoL was normalized in more than one-third of the ITT population by week 12. Achieving a normal QoL is among the goals of CD therapy,1,29 and has been associated with disease activity, among other factors.30
Although the consistency of all measures supports the reliability of the data obtained in this study, limitations should be kept in mind when interpreting these results. This was an open-label study with no control group, and each patient acted as his/her own control. Sample size was somewhat limited, although significant changes were detected in the primary and secondary endpoints. Additionally, a substantial number of patients were lost throughout the study, which led us to include results from both the ITT and the PP populations. Colonoscopy was not used during follow-up and some of the variables used are subjective (QoL measurements), or have been described as prone to subjectivity (HBI). Finally, these results can be only considered for patients with luminal CD.
In conclusion, treatment with adalimumab was associated with a rapid, as early as day 4, clinical response and remission, improvement in QoL and fatigue, and a reduction of inflammatory biomarkers in adult anti-TNF naïve patients with moderately-to-severely active CD. Although these results are encouraging, additional clinical studies are required to further confirm these findings. Time to response is a relevant endpoint for clinical practice in CD, and it is our belief that speed of response should be addressed in future studies.
Authors’ contributionsI. Marín-Jiménez, F. Casellas, M. Barreiro-de Acosta, and M. Esteve were involved in the study concept and design, data collection and analysis, drafting the manuscript, and provided critical edits. L. Castro-Laria, S. García-López, D. Ceballos, A. Echarri, M.D. Martín-Arranz, D. Busquets, J. Llaó, M. Navarro-Llavat, J.M. Huguet, F. Argüelles-Arias, R. Vicente, J.M. Boudet, G. Díaz, and A.M. Sánchez-Migallón were involved in data collection and analysis, drafting the manuscript, and provided critical edits. All authors approved the final version of the manuscript.
FundingFinancial support for the study was provided by AbbVie. AbbVie participated in the study design and conduct, interpretation of data, review, and approval of the publication. No honoraria or payments were made for authorship.
Conflicts of interestI. Marín-Jiménez has served as a speaker, consultant and advisory member for or has received research funding from MSD, AbbVie, Hospira, Takeda, Janssen, Ferring, Faes Farma, Shire Pharmaceuticals, Dr. Falk Pharma, Chiesi, Gebro Pharma, Otsuka Pharmaceuticals, Astrazeneca and Tillotts Pharma. M. Barreiro-de Acosta has served as a speaker, consultant and advisory member for or has received research funding from MSD, AbbVie, Janssen, Kern Pharma, Celltrion, Takeda, Gillead, Pfizer, Ferring, Faes Farma, Shire Pharmaceuticals, Dr. Falk Pharma, Chiesi, Gebro Pharma, Adacyte and Vifor Pharma. M. Esteve has served as a consultant for AbbVie, MSD, Takeda and Tillots Pharma and has received speaker fees from MSD, AbbVie, Janssen y Pfizer. S. García-López has served as a speaker, advisory member for or has received research funding from AbbVie, MSD, Takeda, Janssen, Ferring, Faes Farma, Shire Pharmaceuticals and Chiesi. A. Echarri has received research funding from AbbVie, Janssen and Takeda and speaker fees from AbbVie, MSD, Janssen, Ferring and Pfizer. M.D. Martín-Arranz has received fees as a speaker or consultant, or travel or research grants from MSD, AbbVie, Hospira, Pfizer, Takeda, Janssen, Shire Pharmaceuticals, Tillotts Pharma and Faes Pharma. M. Navarro-Llavat has received research funding from AbbVie and MSD, and speaker fees from AbbVie, MSD, Takeda, Janssen, Ferring, Shire Pharmaceuticals, Zambon, Dr. Falk Pharma, Casen and Allergan. J.M. Huguet has received research funding from AbbVie and MSD and speaker fees from Abbvie, MSD, Ferring, Janssen and Takeda. F. Argüelles-Arias has served as a consultant for AbbVie, MSD, Kern Pharma, Celltrion, and Takeda, and has also received research funding from MSD, Kern Pharma, Celltrion, and Takeda, and speaker fees from MSD, Kern Pharma, Celltrion, and Takeda. R.Vicente has received medical education funding and speaker fees from AbbVie, MSD, Shire, Ferring and Takeda. G. Díaz was AbbVie employee when the manuscript was developed and does not declare another conflict of interest. A. M. Sánchez-Migallón is a full-time employee at AbbVie, Inc. F. Casellas has received research funding from AbbVie, MSD, Shire, Ferring and Zambon. The rest of the authors have no conflicts of interest to declare.
We thank the clinical site investigators Mónica Sierra Ausin, Jesús Barrio Andrés, Pilar Martínez Montiel, Cristina Verdejo Gil, Daniel Carpio López, Ana Guitiérrez Casbas, Eva Iglesias Asenjo, Raquel Camargo Camero, José Manuel Herrera Justiniano, Xavier Aldeguer Manté, Esther García Planella, and Cristina Rodríguez Gutiérrez. Medical writing support was provided by Luis F. García-Fernández, PhD and Blanca Piedrafita, PhD (Medical Statistics Consulting, Valencia, Spain), which was funded by AbbVie.
The following are the supplementary data to this article:
