






Colorectal cancer (CRC) is one of the most important causes of death in the world. Hereditary CRC is found in 5–10% of CRC patients. In this review, we will focus on the major forms of hereditary CRC and their management according to the most recent literature available.
O cancro coloretal (CCR) é uma das mais importantes causas de morte ao nível mundial. O cancro coloretal hereditário está associado a cerca de 5 a 10% de todos os casos de CCR. Neste artigo faz-se uma revisão da abordagem dos principais síndromas hereditários associados a CCR de acordo com a literatura mais recente.
Colorectal cancer (CRC) is one of the most important causes of death in the world. In Portugal, CRC has the second highest incidence after breast cancer in female and prostate cancer in men1 and the second cause of cancer-related death.
The cause of CRC is multifactorial, with inheritance and environment assuming the most relevant roles. Approximately 70–80% of CRC cases seem to be sporadic, while the remaining 20–30% is associated with an inherited pattern. Patients with a familial risk make up approximately 20% of all patients with colorectal cancer, whereas approximately 5–10% of the total annual burden of colorectal cancer is hereditary and Mendelian in nature.
Screening for hereditary cancer syndromes in patients with CRC should include review of personal and family histories and genetic evaluation according to more or less established criteria.
A diagnosis of Lynch syndrome, familial adenomatous polyposis, or another genetic syndrome can influence clinical management of patients with CRC and their family members. A timely identification of individuals at risk for hereditary CRC syndromes offers an opportunity to a sooner intervention or prevention.
In this review we will focus on the major forms of hereditary colorectal cancer, Lynch syndrome, familial adenomatous polyposis, MUTYH polyposis, juvenile polyposis, Peutz–Jeghers syndrome and serrated/hyperplastic polyposis syndrome.
2Lynch syndromeLynch syndrome (LS) is an autosomal dominant condition caused by a defective mismatch repair (MMR) gene.
Although this syndrome has also been known as HNPCC (hereditary non-polyposis colorectal cancer), this terminology is now reserved to patients and/or families who fulfill the Amsterdam criteria. The LS denomination must be only applied to patients and families in which the genetic basis can be linked to a germline mutation in one of the DNA MMR genes or the EPCAM gene.
Lynch-like syndrome patients display alterations in MMR molecular immunohistochemical or microsatellite instability (MSI) without an identifiable germline mutation. Familiar colorectal cancer type X refers to patients that meet Amsterdam I criteria without LS MSI characteristics.
LS is responsible for approximately 3% of all of the newly diagnosed colorectal cancer and is probably the most common hereditary CRC.2 In fact, the major clinical consequence of LS is CRC with a life-time risk varying between 15% and 70% depending on sex and MMR mutated gene. Mean age of CRC diagnosis is 10–15 years earlier than sporadic cases.3
These CRC are predominantly right colon located and have a very rapid adenoma–carcinoma progression, with frequent reports of CRCs arising within three years of a clearing colonoscopy. However, CRC prognosis in LS patients is better when compared to sporadic matched stage CRC.4
The presence of CRC, endometrial, ovary, urinary tract, stomach, small bowel or brain cancer, especially at young ages and with cancer family history, should lead to investigate a probable hereditary cancer. In this clinical setting the genetic counseling has a major role and can include personal and family cancer history, risk assessment, education, informed consent and genetic testing.
Multiple clinical criteria have been developed to identify at risk patients. Obviously, all members of an already known Lynch family should be tested. In individuals without previously Lynch diagnosis, the two most used are Amsterdam criteria (sensitivity 22% and specificity 98%) and Revised Bethesda Guidelines (sensitivity 82% and specificity 77%) but other clinical criteria, like endometrial cancer below 50 years, and computational prediction systems have been applied as well5 (Tables 1 and 2).
Amsterdam I and II criteria for diagnosis of hereditary non-polyposis colorectal cancer.
| Amsterdam I criteria |
|---|
| 1. Three or more relatives with histologically verified colorectal cancer, 1 of which is a first-degree relative of the other two. Familial adenomatous polyposis should be excluded. |
| 2. Two or more generations with colorectal cancer. |
| 3. One or more colorectal cancer cases diagnosed before the age of 50 years. |
| Amsterdam II criteria |
|---|
| 1. Three or more relatives with histologically verified HNPCC-associated cancer (colorectal cancer, cancer of the endometrium, small bowel, ureter, or renal pelvis), 1 of which is a first-degree relative of the other 2. Familial adenomatous polyposis should be excluded. |
| 2. Cancer involving at least 2 generations. |
| 3. One or more cancer cases diagnosed before the age of 50 years. |
Revised Bethesda guidelines.
| 1. CRC diagnosed at younger age than 50. |
| 2. Presence of synchronous or metachronous CRC or other LS-associated tumors. |
| 3. CRC with MSI-high pathologic-associated features (Crohn-like lymphocytic reaction, mucinous/signet cell differentiation, or medullary growth pattern) diagnosed in an individual younger than 60 years old. |
| 4. Patient with CRC and CRC or LS-associated tumor diagnosed in at least 1 first-degree relative younger than 50 years old. |
| 5. Patient with CRC and CRC or LS-associated tumor (colorectum, endometrium, stomach, ovary, pancreas, ureter, renal pelvis, biliary tract, brain, small bowel, sebaceous glands, and kerotoacanthomas) at any age in 2 first-degree or second-degree relatives. |
Patients meeting Amsterdam criteria should undergo direct germline testing. On the other hand, for those who meet Revised Bethesda criteria, evaluation by immunohistochemical testing for the MLH1/MSH2/MSH6/PMS2 proteins and/or testing for microsatellite instability is suggested.
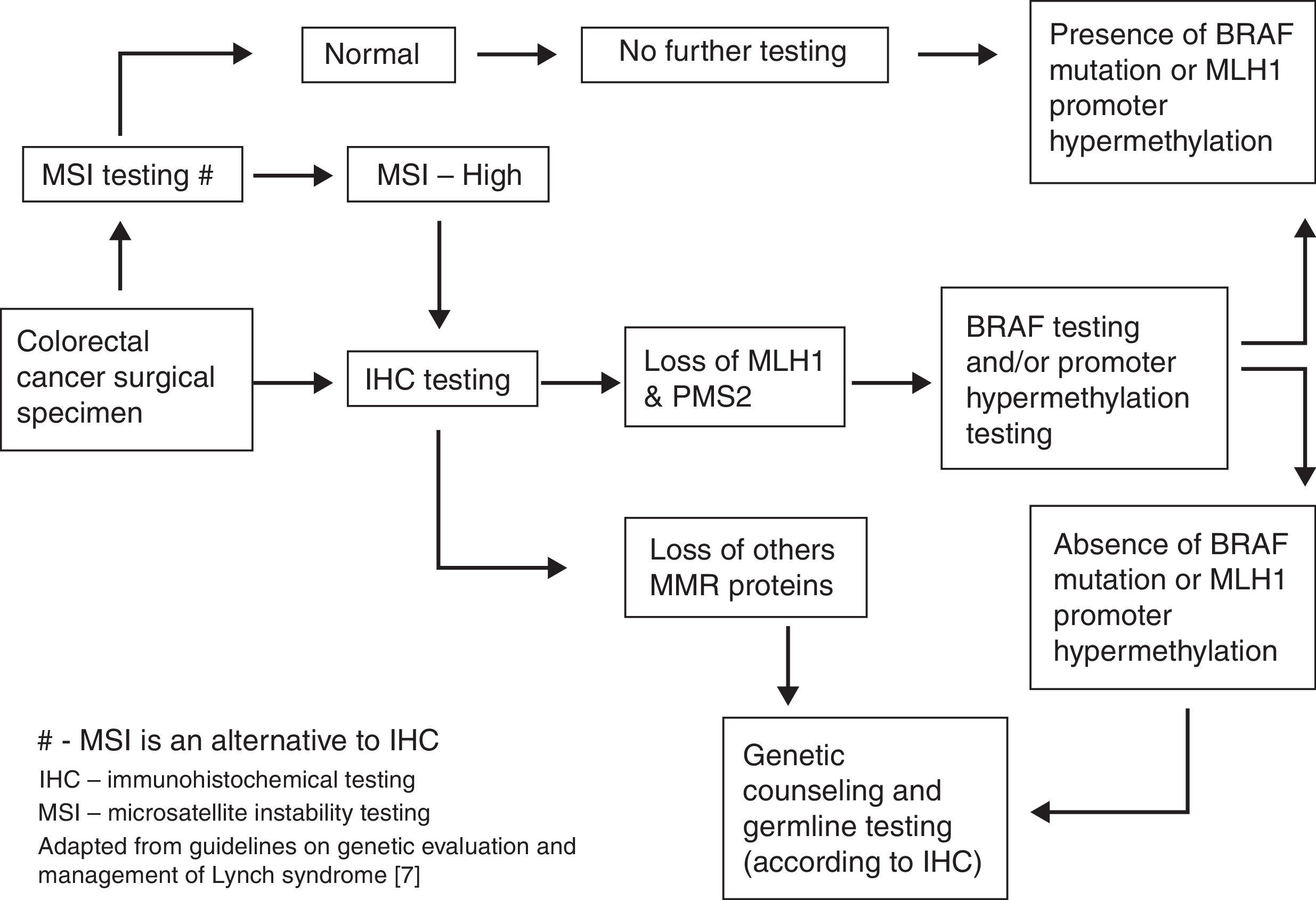
Universal testing for all newly diagnosed CRC (or CRC patients under 70 years old) is currently a hot topic under discussion (Fig. 1). In this setting, tumor immunohistochemistry testing seems to be more sensitive and cost-effective for identifying LS patients and achieves the aim of reduced morbidity and mortality. However, implementation of this screening system is complicated and requires effective multidisciplinary approach.6–9

As long as the clinical criteria to search LS are fulfilled, different options can be adopted for detecting MMR defect.
Tumor testing can be done on archived formalin-fixed tissue for surgical resection or biopsies specimens. Microsatellite instability testing (sensibility 85%, specificity 90%) or preferably immunohistochemistry testing of tumor tissue for searching the lack of expression of MMR gene proteins (sensibility 83%, specificity 90%) can be done according to local resources and expertise.
The lack of a specific MMR repair gene in IHC (MLH1, MSH2, MSH6, PMS2) can indicate germline testing to that specific gene. However, in case of loss of MLH1/PMS2 protein expression in the tumor, analysis of BRAF V600E mutation or analysis of methylation of the MLH1 promoter should be carried out first to rule out a sporadic case. If loss of any of the other proteins (MSH2, MSH6, PMS2) is observed, germline genetic testing should be done for the genes corresponding to the absent proteins (MSH2, MSH6, EPCAM, PMS2, or MLH1).
When tumor tissue is not available, we should consider direct genetic testing. Full germline genetic testing for Lynch syndrome should include DNA sequencing and large rearrangement analysis.10
Germline testing for deleterious mutation in MLH1, MSH2, MSH6, PMS2 and EPCAM can confirm LS diagnosis, to establish at risk status of family members and allow an adequately planning of their management. Also, the prompt diagnosis of these patients facilitates their surgical approach.
In a LS mutation known family, mutation absence in an individual is considered a negative test and its presence is a positive test leading to a surveillance plan implementation. When mutation is not known in LS pedigree, at risk individuals should be managed as a positive test and undergo periodic assessments as new genetic data emerge.
LS patients are at increasing risk of colorectal and extracolonic cancers at young age but there are several clinical differences according to the gene mutated.
MLH1-mutation carriers tend to develop CRC at younger ages, whereas MSH2 carriers seem to be at higher risk for extracolonic cancers. MSH6 mutations female carriers have an increased risk for endometrial cancer which may surpass the lifetime CRC risk. In contrast, the risks for CRC and endometrial cancer seem to be lower among individuals with mutations in PMS2 compared with other MMR gene mutations.
Current guidelines do not tailor recommendations according to each genetic defect and suggest surveillance beginning at 20–25 years, including clinical history, physical examination and patient and family education.
Potential psychosocial problems related to genetic testing and surveillance must be monitored and prompt referral to a clinical psychologist should be done if increased psychological stress is detected.11
CRC is the major consequence of LS syndrome and colonoscopy screening is the only measure associated to a decreasing CRC incidence and mortality.12,13 Almost all societies and multitask forces recommend total colonoscopy for at-risk persons or LS patients, every 1–2 years beginning at 20–25 years or 2–5 years before the youngest case in the family if CRC diagnosis before 25 years old.13
In patients with MSH6 and PMS2, surveillance could start at 30 or 35 years respectively, given the later age of CRC diagnosis. The screening program should continue until age 70–75 or comorbidity.
The second most important cancer in Lynch Syndrome is endometrial cancer with a cumulative lifetime risk up to 60%. Several modalities of screening have been debated but none of these show benefits in survival. Most societies and multitask forces suggest annual pelvic examination and endometrial sampling starting at age 30–35 years.14,15
As the endometrial cancer, ovarian cancer screening does not have a survival impact. However, annual transvaginal ultrasound starting at the same age is suggested.7
Urinary tract cancer in LS patients has an estimate lifetime risk up to 20%. There is no evidence of screening benefits. Urinary cytology is one of the most widely used screening approaches, but the lack of sensitivity and the many false positive results requiring invasive procedures led to an abandon of this attitude in clinical practice. Urinalysis is accessible and non-expensive and may be annually considered in LS patients at age 30–35 years.7
The gastric cancer in LS has a lifetime risk around 8%. The majority of these cancers are intestinal and amenable to endoscopic surveillance. Esophagogastroduodenoscopy (EGD) with gastric biopsy should be done at age 30–35 years with Helicobacter pylori treatment if applicable. Subsequent endoscopic surveillance may be considered every 2–3 years.11,16
No current evidence exists to support routine screening of small bowel cancer in LS patients. The majority of these cancers seem to be located in the duodenum or ileum and within the reach of EGD and colonoscopy with ileal intubation which may be the only reasonable approach.17
The pancreatic screening is still being debated but almost no society recommends this practice. It may only be considered if a pancreatic cancer diagnosis exists in a first degree relative.18
Total colectomy with ileorectal anastomosis is the standard treatment in LS patients with colon cancer or colorectal lesions not removable by endoscopic therapy. The high rate of metachronous CRC in LS patients with segmental resection supports this approach. For patients not amenable for colorectal screening this surgical option may be considered. After colectomy, flexible sigmoidoscopy every two years is recommended by some societies.16,19
Hysterectomy and bilateral salpingo-oophorectomy can be recommended. All pros and cons of prophylactic gynecological surgery should be discussed in LS women who have finished childbearing or at the age of 40. Patient considerations in this decision could include uterine cancer risk depending on MMR gene mutation, morbidity of surgery and the risk of menopausal symptoms. If CRC surgery is scheduled, the option of prophylactic surgery at the same time should be considered.20,21
Some studies suggest that aspirin can reduce incidence of colorectal and extracolonic cancers. This approach can be discussed with patients bearing in mind patient-specific risks, benefits, and uncertainties of treatment, but no strong evidence exists to support this practice as a formal recommendation.22–24
Also, patients could be advised to stay within the normal weight range and avoid smoking.11
3Familial adenomatous polyposisFamilial adenomatous polyposis (FAP) is an autosomal dominantly inherited syndrome that arises from a germline mutation on APC tumor suppressor gene with a nearly 100% penetrance. The most important clinical features are the presence of hundreds to thousands adenomas throughout the colorectum at an early age and a lifetime risk for colorectal cancer close to 100%. FAP accounts for approximately 1% of all cases of colorectal cancer.25
Patients with FAP can also developed benign extracolonic manifestations as fundic gastric polyps, desmoid tumors, cutaneous lesions, osteomas, odontomas, adrenal adenomas and pigmented ocular fundic lesions. The second most important cancer is duodenal cancer (4–12%) but hepatoblastoma (1–2% at age five), thyroid (<2%), pancreatic, brain and biliary tree cancer can also occur.
Some variants of FAP are known by specific clinical features like Gardner syndrome (sebaceous cysts, osteomas, dental abnormalities), Turcot syndrome (medulloblastomas) and attenuated FAP. Attenuated FAP is characterized by right colon predominance oligopolyposis and typically delayed CRC, arising from a mutation at the extreme 3′ or 5′ end of the APC gene.
Despite the selective disadvantage of the disease, the incidence of FAP is maintained by the frequency of new mutation which may reach one quarter of patients. In these cases the clinical suspicion is essential for diagnosis and genetic testing.26
All patients with a FAP first-degree relative should be proposed to APC gene testing at 10–12 years, as well as all individuals presenting classic PAF phenotype.
For patients with 10 or more cumulative colorectal adenomas the genetic testing should also be done. Genetic counseling is an essential part of genetic testing.
The screening program should begin at 10–12 years old in a patient with a mutation positive test or if the patient is a first degree relative of a PAF patient without a known mutation, which account to nearly 20% of the cases.
Annual sigmoidoscopy is recommended until appearance of colorectal adenomas. After that, annual colonoscopy should be performed until the late adolescence or appearance of advanced lesions not amenable to therapeutic endoscopy that lead to an earlier surgical approach.
Several programs of surveillance have been proposed for patients with familiar PAF history without identified mutation Most of these suggest annual sigmoidoscopy beginning at 10–12 until 25 years and bi-annual thereafter until 30. After that age, in the absence of colorectal lesions, the sigmoidoscopy could be done every three years and after the age of 50, the approach could be done as an average risk colorectal cancer patient.16,27
After that age, in the absence of colorectal lesions, the sigmoidoscopy could be done every three years and after the age of 50, individuals could be managed as average risk colorectal patients.
Currently, chemoprophylaxis is not recommended as a primary approach.28
Total colectomy is the only definitive approach to prevent CCR in FAP patients. Although most PAF patients were proposed to a surgical approach between 16 and 25 years old, this timing should be individualized according to number and histologic features of polyps, family history of early cancer or genetic disposition.16
Prophylactic surgical options are either colectomy and ileorectal anastomosis (IRA) or proctocolectomy and ileal pouch-anal anastomosis (IPAA).29,30
Both surgical techniques have pros and cons namely surgical complexity, preservation on sphincter function and fertility, quality of life, postsurgical endoscopic surveillance timings and CCR risk.
IRA is technically straightforward and has a low complication rate, namely sexual or bladder dysfunction. However, patients who undergo colectomy with IRA are at a 25% risk of developing cancer in the retained rectum after 20 years.31
IPAA is preferable in extensive rectal polyposis, curable rectal cancer or in patients not reliable for remaining rectum surveillance. Recent studies also favor IPAA approach in view to reduce CCR risk.32,33
Although total proctocolectomy with permanent end ileostomy removes the risk of CCR cancer, the inevitable and definitive stoma limits this approach to patients not suitable for anastomotic options or locally advanced CCR.
After colectomy, the remaining rectum, ileal pouch or terminal ileum has to be addressed.
In IRA approach, it may be reasonable to begin rectal endoscopic surveillance six months after surgery and then once a year. The initial surveillance can be the same for IPAA or terminal ileostomy but further endoscopic evaluations can be extended to every 2–3 years. Polyps found should be endoscopically removed, if possible, prompt reoperation should be planned in case there is a diagnosis of at least CCR T1.
Postsurgical chemoprophylaxis can be suggested but, nowadays, it is not known if polyps’ reduction decreases cancer risk. Furthermore, the benefits of these agents in long-term use need to be closely weighed against the risk of potential gastrointestinal and cardiovascular side effects.28,34,35
For upper gastrointestinal cancer most societies recommend upper endoscopy every two years from the age of 25. Axial endoscopic view allows an observation of gastric and duodenal lesions but for Vater ampulla definition, it may be more adequate with lateral endoscopic view. Chromoendoscopy with indigo carmine dye may be considered.36
There is no clear evidence to support screening for gastric cancer in FAP patients. However, given the increased risk for duodenal cancer, the stomach should be examined at the same time of duodenoscopy.
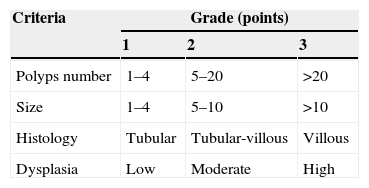
In Spigelman classification, four stages are defined according to number, size and histology features of duodenal polyps, which allow us to decide future follow-up and therapeutic approach (Table 3).37
The next upper endoscopy surveillance is recommended in 5 years for stage 0–I; 3 years for stage II, 1–2 years for stage III and every six months for stage IV.
In stage 0-II patients, neither chemoprophylaxis nor surgical approach are indicated. In stage II–III only chemoprophylaxis is suggested. The therapeutic role of endoscopy is not yet established in these patients but it may delay a stage progression. In stage IV patients, surgical approach (Whipple or duodenectomy if possible) is consensual but new therapeutic endoscopic techniques are emerging in this setting.38
Enteroscopy capsule or small bowel radiographic contrast study started at the age of 20 or, eventually, preoperative enteroscopy at the colectomy time can be suggested for small bowel evaluation, according to local resources and expertise, but more studies are needed in this setting.39–41
Adequate physical examination for searching abdominal masses, namely desmoids tumors, is essential. Abdominal ultrasound, CT scan or RNM may be helpful, especially in patients with familial history, starting 1–2 years after colectomy and then every 5–10 years.
If intra-abdominal or abdominal wall desmoids are detected, medical therapeutic with sulindac and tamoxifen can be suggested. Surgery, chemotherapy (doxorubicine and dacarbazine or methotrexate and vinblastine) or radiation therapy are also available options.42
Thyroid physical evaluation and complementary US should begin during adolescence.43
For children with affected parents, hepatoblastoma should be screened until 5–7 years old. For pancreatic lesions no additional tests are recommended.27
For attenuated PAF, surveillance options are different according to specific clinical features. Total colonoscopy should be the initial endoscopic option starting at late teenage years and then every 2–3 years until adult age. Thereafter, yearly total colonoscopy should be done until important polyposis occurs and colectomy proposed.16 If no mutation is found surveillance should be maintained until 75 years old or comorbidities.16
If Gardner syndrome is detected, the specific surveillance is limited to the early detection of osteomas and dental abnormalities. In Turcot syndrome, regular brain tomography for meduloblastoma detection may be considered.
For these three specifics phenotypes the remaining surveillance is similar to classic PAF.
4MUTYH associated polyposisMUTYH associated polyposis (MAP) is the only autosomal recessive polyposis syndrome, caused by biallelic mutations in the MUTYH gene.44 Because the diagnostic criteria for MAP are yet to be established, it is difficult to diagnose the disease. Furthermore, the clinical features of MAP may vary. Although with a clinical presentation similar to attenuated FAP, it is now clear that the clinical spectrum of MUTYH germline mutations is broad and can include CRC without polyposis or even overlapping classic FAP.45 In this syndrome, adenomatous polyps are the most common CRC but serrated polyps are also common.46
Although the increased CRC risk in patients with biallelic mutations is well established, there is some controversy regarding individuals with monoallelic mutations.47
Extracolonic disease may include gastric and duodenal polyps, duodenal carcinoma, osteomas and dental cyst, breast cancer in women, congenital hypertrophy of the retinal pigment epithelium and sebaceous gland tumors (Muir-Torre phenotype). There also appears to be an increased risk of ovarian and skin cancer.48
MUTYH screening should be directed to patients with more than 10 adenomas and/or hyperplastic/serrated polyps, especially in the context of a family history with recessive inheritance pattern.49 Biallelic MUTYH mutations are found in about 28% of APC mutation-negative patients with 10–100 polyps and in 14% of patients with more than 100 polyps.50 Patients without Lynch syndrome and a cumulative number of adenomas between five and nine should also undergo screening for MUTYH, in the presence of an appropriate setting: less than 40 years old, at least five advanced adenomas, association with sebaceous neoplasms or duodenal polyposis.51
Until this day, there are no widely accepted screening guidelines for these patients. Biallelic MUTYH mutation positive patients, or a not tested sibling of a patient with MAP, should start colonoscopy at the age of 25–30 and repeat every 2–3 years if normal. If polyps are found, the next colonoscopy should be in 1–2 years.16
However, an earlier colonic surveillance, at age 20 years, is also suggested by some societies and expert panels.49,52
Although colectomy may be considered at the age of 21 years, surgery timing should be individualized according to polyposis features and therapeutic endoscopic possibility. The surgical approach must take into account rectal polyp burden.
Upper gastrointestinal endoscopy (including duodenoscopy) should be considered every 3–5 years, beginning at least at age 30–35 years.16,52
Women with biallelic MUTYH gene mutations may be consider to have a high-risk breast cancer and should be advised for adequate surveillance. At least one dermatological observation at the diagnosis must be performed, and patients should be aware to identify new skin lesions.51
CRC risk associated with monoallelic MUTYH carriers is still under debate. To date CRC screening, as recommended for first-degree relatives of a patient with sporadic CRC, is advised.52
5Peutz–Jeghers syndromePeutz–Jeghers syndrome (PJS) is a rare autosomal dominant disorder that is characterized by multiple gastrointestinal hamartomatous polyps and lips and buccal mucosa pigmentation.53 Prevalence is estimated to be 1:100,000–1:200,00054 and the diagnosis is often made during the second decade, with a median age of 11 years old.55 The most common and known cause is a combination of a first allele germline mutation of tumor supressor gene STK11 with a somatic one of the second allele.56
The two main aspects in the management of PJS patients are the long term cancer risk and PJ polyps related complications, such as intussusception and bleeding.57 Individuals have an increased risk for gastrointestinal and non-gastrointestinal neoplasms. Lifetime cumulative risk for all cancers is up to 90%; most of them are colorectal, breast, gastric and pancreatic cancers, but other tumors have been associated with PJS (Table 4).16,58,59
Clinical diagnostic criteria have been established in 2010 by World Health Organization and revised by an European expert consensus (Table 5).60
Clinical diagnostic criteria for PJS.
| Suggestive family history of Peutz–Jeghers syndrome AND… | Any number of PJ polypsORCharacteristic mucocutaneous pigmentation |
| Non-suggestive family history AND… | Two or more histologically confirmed PJ polypsORAny number of PJ polyps in the presence of characteristic mucocutaneous pigmentation. |
Individuals who meet clinical criteria for PJS should undergo genetic testing for a germline mutation in the STK11 gene to confirm diagnosis and counsel family members. If no pathogenic STK11 mutation is found but the individual meets clinical criteria for PJS, the clinical criteria prevail over genetic test, since the diagnosis is not excluded because not all mutations responsible for PJS are identified.61 In families with an unknown mutation it is necessary to search those who develop early SPJ clinical signs and then offer them appropriate surveillance.61
None of the screening recommendations have been validated, but some groups of experts have proposed surveillance recommendations.
Endoscopic surveillance may include62 a first upper and lower gastrointestinal endoscopy at eight years old. If polyps are present, the surveillance should be repeated every three years; if not, the second endoscopic examination can be done at age 18 and then every three years. After the age of 50 years, colonoscopy should be done not three but every one to two years. Also at age eight years, video capsule endoscopy should be considered, and the same intervals as for upper and lower gastrointestinal endoscopies apply.
All patients with Peutz–Jeghers syndrome should be screened for pancreatic cancer, regardless the family history. Suggestions for Initial approach include endoscopic ultrasonography and/or magnetic resonance cholangiopancreatography which can begin at age 25 years and then every one to two years.17,60
Annual breast MRIs are recommended, starting by the age of 25 years and regular clinical breast examination should also be performed.16
For men, annual testicular examination starting at age of 10 years is recommended.16
There is controversy regarding the gynecological cancers screening but we can consider CA-125 blood test and transvaginal ultrasound.16,60,62
No specific recommendation for lung cancer screening has been made. However, education about smoking cessation should be performed.16
6Juvenile polyposis syndromeJuvenile polyposis syndrome (JPS) is a rare (<1:100,000) autosomal dominant disease with high penetrance, characterized by the occurrence of juvenile polyps in the gastrointestinal tract and an increased risk of colorectal cancer.63 JPS is associated with germline mutations in three genes (SMAD4, BMPR1A and ENG), all related to the TGF-b pathway. In patients fulfilling the diagnostic criteria, it is possible to detect mutations in only approximately 50%.64
Juvenile polyposis syndrome is defined by the presence of five or more juvenile polyps in the colon, multiple juvenile polyps found throughout the gastrointestinal tract or any number of juvenile polyps in an individual with a positive family history of juvenile polyposis.16 A family history of juvenile polyps is found in 20–50%,65 and those are called familial juvenile polyposis.
The disease has been phenotypically classified into three subsets, but these forms appear to be variable expressions of the same disease.63 The age at diagnosis varies, but symptoms are usually present in the first and second decades of life.65 Typical presenting symptoms include rectal bleeding, anemia, abdominal pain, diarrhea, and mechanical complications such as intussusception, obstruction, and polyp prolapse.65
An increased cumulative risk for both colorectal cancer (38%) and upper GI cancer (21%) has been documented.64 Pancreatic cancer and small bowel cancer have also been reported.63
Prophylactic total or subtotal colectomy or gastrectomy should be considered in patients with multiple polyps, severe symptoms or a family history of CRC.64 Proctocolectomy and subtotal colectomy with ileorectal anastomosis need endoscopic follow-up because of the high recurrence-rate of polyps.
There is limited data and therefore no wide consensus has been established for screening or for the surveillance and management of patients with clinical diagnostic features of juvenile polyposis.
For asymptomatic at-risk members of JPS families British recommendations state surveillance with colonoscopy every one to two years starting at age 15–18 years until age 70 and gastroduodenoscopy starting at age 25 and one to two year interval thereafter.66 Small-bowel disease is not a significant clinical problem in JPS and surveillance should not be performed.67 There are no suggestions of pancreatic screening modalities.
7Serrated polyposis syndromeSerrated polyposis syndrome (SPS) is a rare condition characterized by a predisposition to serrated polyps and an increased risk for colorectal cancer and possibly some other extracolonic neoplasms.
In contrast to FAP and Lynch syndrome, no genetic abnormality has been consistently described in SPS, but inheritance is seen in a small percentage of families.68
Although routine germline testing is not routinely recommended for SPS patients, MUTYH testing may be considered if concurrent adenomas and/or a family history of adenomas are present.69
SPS is generally described as the presence of multiple, large and/or proximal hyperplastic/serrated polyps.70 Serrated polyps, as characterized by the saw-toothed architecture, comprise heterogeneous lesions, including hyperplastic polyps, sessile serrated adenomas, traditional serrated adenomas and mixed lesions of these.71–73
Clinical criteria for diagnosis were defined by the World Health Organization and include at least one of the following: at least five serrated polyps proximal to sigmoid colon with ≥2 of these being >20mm, any number of serrated polyps proximal to the sigmoid colon in an individual who was a first-degree relative with serrated polyposis and >20 serrated polyps of any size distributed throughout the colon.74
There are no available studies regarding the effectiveness of surveillance in SPS. However, based on cancer risk, these patients should undergo colonoscopies every 1–3 years with attempted removal of all polyps or, at least, all polyps bigger than 5mm.16,75
The inability to control polyps growth constitutes an indication to colectomy with IRA.
Screening recommendations for individuals from SPS families are not yet established but it is reasonable to screen first degree relatives based on results of baseline evaluations in family members.9
8ConclusionIn summary, it is important to be aware of the possibility of CRC associated hereditary syndromes. A timely identification of these syndromes is a unique opportunity to a sooner and efficient CRC prevention and management.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that no patient data appear in this article.
Right to privacy and informed consentThe authors declare that no patient data appear in this article.
Conflicts of interestThe authors have no conflicts of interest to declare.
