


Refractory celiac disease is an uncommon but serious complication of celiac disease. We describe a case of a severe refractory celiac disease type II, complicated with ulcerative jejunoileitis, in a 68 years old female, unresponsive to consecutive treatments with budesonide, prednisolone, cladribine and autologous stem cell transplantation. The patient maintained severe malnutrition, advanced osteoporosis, anaemia, vitamin deficiencies and hydro-electrolytic imbalances, necessitating consecutive hospitalizations for total parenteral nutrition. The patient also developed life-threatening complications, namely respiratory and urinary septic shock and also episodes of haemorrhagic shock secondary to ulcerative jejunoileitis. The progression to enteropathy associated T-cell lymphoma was never demonstrated, but the patient died 7 years after the diagnosis due to a septic shock secondary to a nosocomial pneumonia and osteomyelitis related to a spontaneous hip fracture. This case highlights the difficulties in the diagnostic process, therapeutic management and surveillance of this rare condition associated with very poor prognosis.
A doença celíaca refratária é uma complicação rara mas muito severa da doença celíaca. Apresentamos o caso de uma doente de 68 anos, com doença celíaca refratária tipo II complicada de jejunoileíte ulcerativa, que não respondeu a tratamentos sucessivos com budesonido, prednisolona, cladribina e transplante autólogo de medula óssea. A doente manteve desnutrição e osteoporose severas, deficiências vitamínicas e desequilíbrios hidro-eletrolíticos, necessitando de múltiplas hospitalizações para receber nutrição parentérica total. Também desenvolveu diversas complicações potencialmente fatais nomeadamente sépsis com origem respiratória e urinária e choque hemorrágico secundário à jejunoileíte ulcerativa. Contudo, nunca se demonstrou a presença de um linfoma de células T. A doente faleceu 7 anos após o diagnóstico devido a choque sético secundário a pneumonia nosocomial e osteomielite relacionada com fratura espontânea da anca. Este caso ilustra as dificuldades sentidas no decurso do diagnóstico, terapêutica e vigilância desta entidade clínica rara, a qual está associada a um péssimo prognóstico.
Celiac disease (CD) is a chronic immune-mediated enteropathy precipitated by exposure to dietary gluten in genetically predisposed individuals. It is a common autoimmune disease affecting around 1% of the general population.1 The diagnosis is often easy in the presence of classic symptoms of malabsorption, positive celiac-specific antibodies and villous atrophy in duodenal biopsies. However, some patients, mainly those diagnosed in the adulthood, can only present atypical or extra intestinal manifestations (e.g. iron deficiency, fatigue, abnormal liver function tests, bone disease, skin disorders, peripheral neuropathy, depression), and the specific serology can be negative.2,3 Strict life-long gluten free diet (GFD) is the only treatment available, leading to clinical and mucosal recovery in most patients, but a small minority does not show clinical improvement upon GFD.1,4 Refractory CD (RCD) is a rare and severe condition, developing in 1–5% of the adult-onset CD patients, and is defined as persistent or recurrent symptoms and signs of malabsorption with small intestinal villous atrophy despite a strict GFD for more than 12 months in the absence of other causes of villous atrophy.5,6 RCD is divided in two types based on the absence (Type I) or presence (Type II) of abnormal intraepithelial lymphocytes (IEL).7 Type II RCD (RCD-II) has limited therapeutic options, often complicates with severe malnutrition, ulcerative jejunoileitis and enteropathy associated T-cell lymphoma (EATL).7,8
We report an unfavourable case of RCD-II, describing the difficulties in the initial diagnosis, the consecutive failures of distinct therapeutic options, and the multiple complications that finally led to the death of the patient.
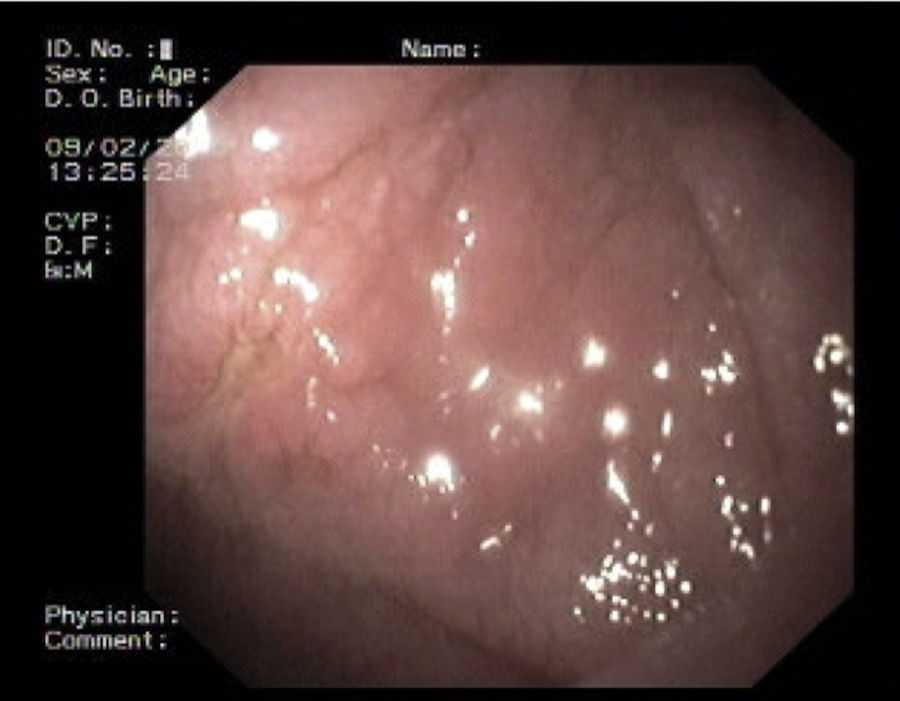
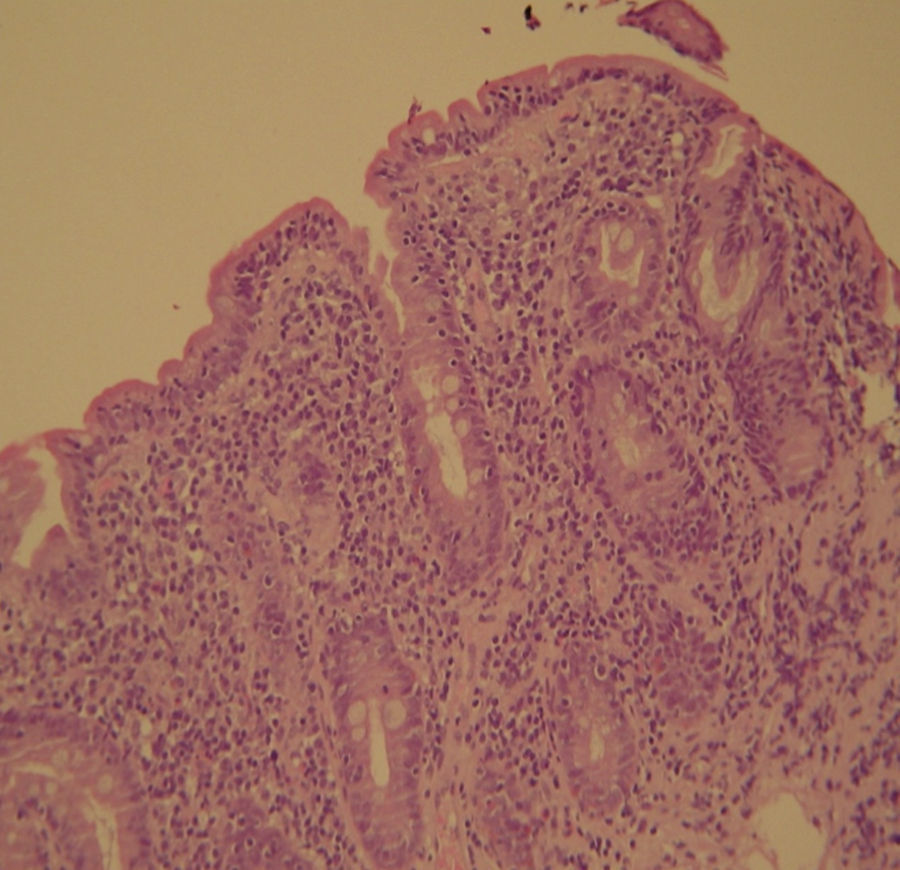
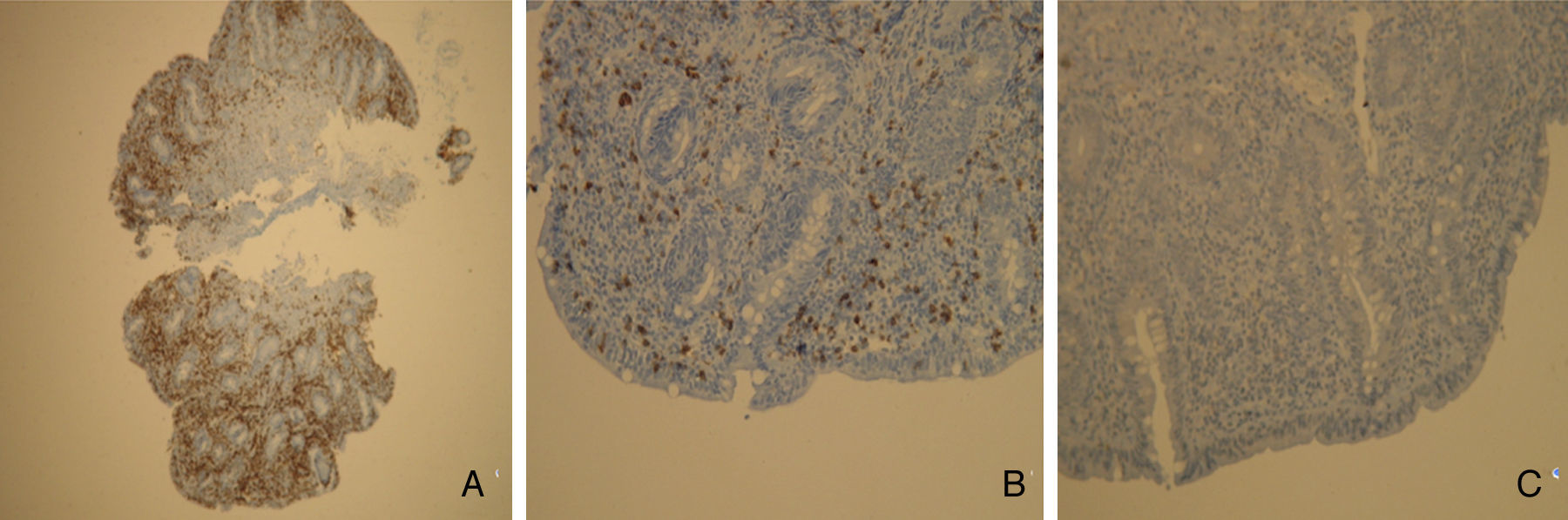
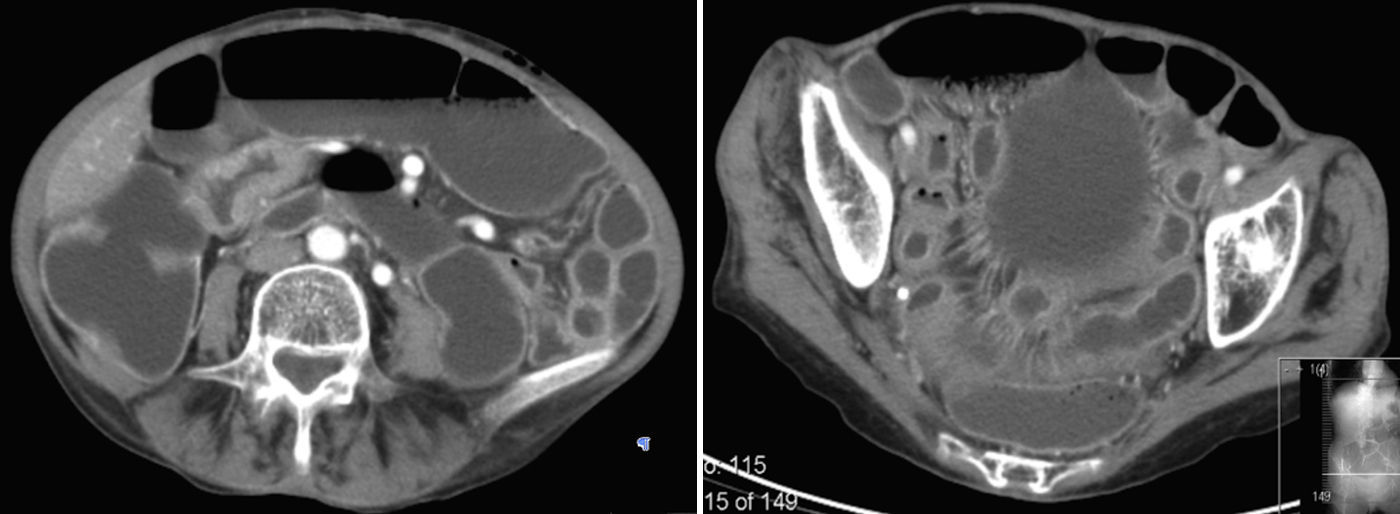
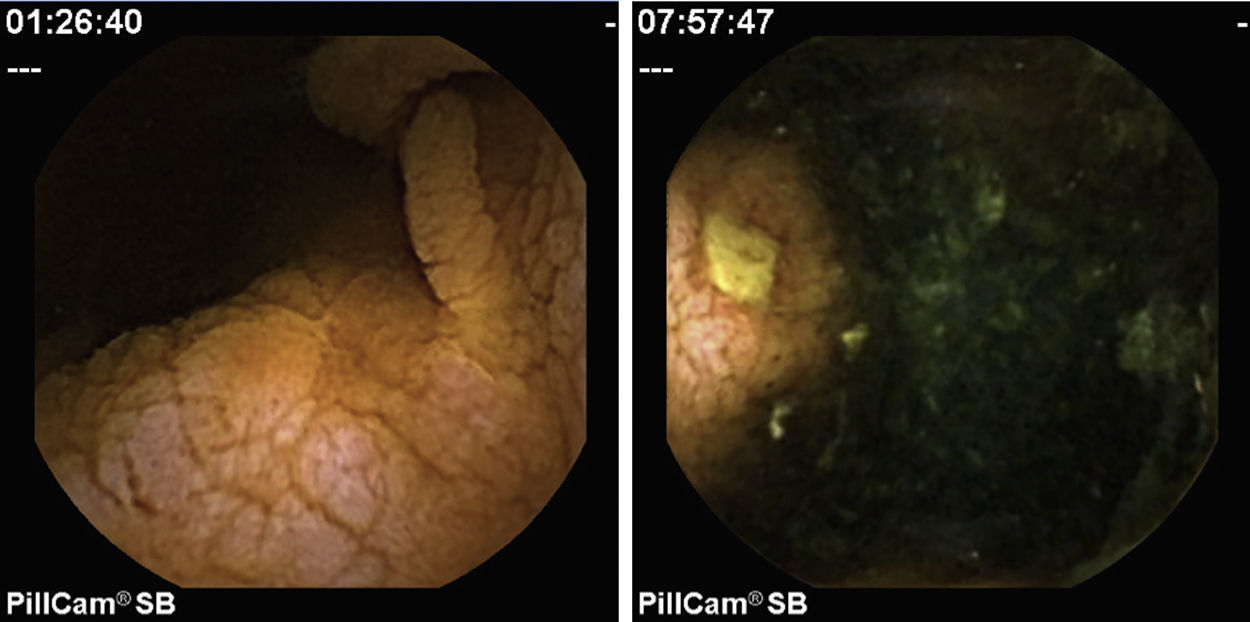
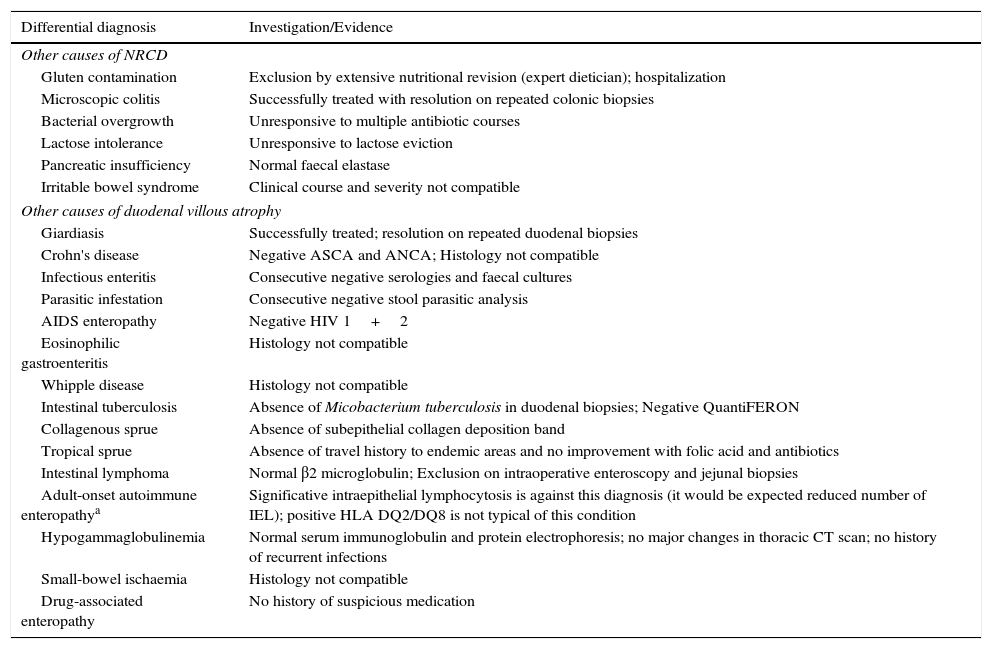
2Case reportWe present the case of a 68 years old white female, diagnosed with CD in 2008, when she was 61 years old, and whom never become asymptomatic despite strict GFD. The diagnosis was made considering characteristic histologic features, despite negative specific-CD serology. She had no other comorbidities apart from chronic depression and there was no family history of CD or other autoimmune diseases. The patient presented to our department for the first time in February 2010 complaining of chronic diarrhoea, vomiting, anorexia, asthenia and weight loss, despite 2 years of GFD. She showed severe malnutrition (body mass index – BMI: 12kg/m2, extreme hypoalbuminemia), threatening hydro-electrolytic imbalances (hypomagnesaemia, hypokalaemia, hypophosphatemia, hypocalcaemia), normocytic anaemia (haemoglobin of 8.1g/dL), coagulopathy (INR of 8.2), vitamin deficiencies and severe osteoporosis. The adherence to GFD was revised and ensured. Upper gastrointestinal (UGI) endoscopy with duodenal biopsies was repeated, showing endoscopic aspects compatible with CD (Fig. 1). A marked villous atrophy, with villous to crypt ratio of <1/1 and increased IEL were observed – Marsh-Oberhuber stage 3b (Fig. 2). These biopsies also allowed the diagnosis of giardiasis that was successfully treated with metronidazole (confirmed with repeated biopsies after treatment). Ileocolonoscopy was performed, and despite absence of mucosal changes random biopsies demonstrated the presence of lymphocytic colitis. Apart from these two parallel diagnoses that could contribute to the refractory symptoms, careful investigation and exclusion of other entities that could be the cause of symptoms and villous atrophy was made (Table 1). Specific-CD serology was repeated and it was negative once again. HLA typing was determined and revealed a DQ2/DQ8 compound heterozygote. Immunohistochemical analysis was performed in duodenal biopsies and revealed more than 50% of aberrant IELs, which were CD3+, but mostly CD8- and CD30- (Fig. 3), being compatible with a RCD-II, with no evidence of EATL. The aberrant pattern was confirmed by detection of T-cell receptor chains clonal rearrangement, performed at the Celiac Centre Amsterdam, VU University Medical Centre. A CT scan enteroclysis identified bowel-wall thickening in the jejunum and ileum, mesenteric lymphadenopathy and prominent mesenteric vessels resembling the comb sign (Fig. 4). Unfortunately, the double ballon enteroscopy performed was not satisfactory and could not find these abnormalities. Additionally, the videocapsule enteroscopy revealed flat mucosa, scalloping effect and multiple small ulcers, particularly in the jejunum (Fig. 5). Apart from total parenteral nutrition (TPN) with correction of ionic and vitamin deficiencies, and after antibiotic therapy to treat the giardiasis, the patient started budesonide (9mg/i.d.) that was adequate not only for the RCD but also for the lymphocitic colitis treatment, yet due to insufficient improvement prednisolone was initiated (1mg/kg weight). The clinical response was unsatisfactory and consecutive hospitalizations were necessary to perform TPN and hydro-electrolytic corrections. Due to lack of response to budesonide and prednisolone an intraoperative enteroscopy was performed and an ulcerative jejunoileitis was identified, corroborating the diagnosis of RCD-II. The case was discussed and reviewed by experts in CD at Celiac Centre Amsterdam, VU University Medical Centre that agreed with the diagnosis of RCD-II and suggested treatment with cladribine, which was performed in October 2010 (single cycle, intravenous, 0.1mg/kg/day for 5 days), with good clinical, analytical and nutritional response (BMI: 20kg/m2), keeping, however, a Marsh-Oberhuber stage 3b. In April 2011, 6 months after cladribine, there was a new clinical deterioration and TPN was again initiated. The patient was then proposed for autologous stem cell transplantation (ASCT), performed in October 2011, with no histological improvement but with some clinical stability until December 2013. Since then the patient was consecutively hospitalised due to severe malnutrition (BMI<14kg/m2), vitamin deficiencies, hydro-electrolytic imbalances and anaemia, necessitating TPN and blood transfusions, and also respiratory and urinary infections complicated with septic shock, and episodes of haemorrhagic shock. There was also a spontaneous fracture of the femur neck, secondary to severe osteoporosis, treated with a dynamic hip screw (DHS) that infected subsequently and needed to be removed. The patient died 7 years after the diagnosis of CD, due to a respiratory septic shock, presenting with severe malnutrition (BMI 10kg/m2), without evidence of EATL on CT scan enteroclysis and PET scan.

 showing severe villous atrophy together with intraepithelial lymphocytosis (>40 IELs/100 enterocytes) and crypt hyperplasia – Marsh-Oberhuber type 3b.")
Other causes of non-responsive celiac disease (NRCD) and villous atrophy in adults that were investigated/excluded.
| Differential diagnosis | Investigation/Evidence |
|---|---|
| Other causes of NRCD | |
| Gluten contamination | Exclusion by extensive nutritional revision (expert dietician); hospitalization |
| Microscopic colitis | Successfully treated with resolution on repeated colonic biopsies |
| Bacterial overgrowth | Unresponsive to multiple antibiotic courses |
| Lactose intolerance | Unresponsive to lactose eviction |
| Pancreatic insufficiency | Normal faecal elastase |
| Irritable bowel syndrome | Clinical course and severity not compatible |
| Other causes of duodenal villous atrophy | |
| Giardiasis | Successfully treated; resolution on repeated duodenal biopsies |
| Crohn's disease | Negative ASCA and ANCA; Histology not compatible |
| Infectious enteritis | Consecutive negative serologies and faecal cultures |
| Parasitic infestation | Consecutive negative stool parasitic analysis |
| AIDS enteropathy | Negative HIV 1+2 |
| Eosinophilic gastroenteritis | Histology not compatible |
| Whipple disease | Histology not compatible |
| Intestinal tuberculosis | Absence of Micobacterium tuberculosis in duodenal biopsies; Negative QuantiFERON |
| Collagenous sprue | Absence of subepithelial collagen deposition band |
| Tropical sprue | Absence of travel history to endemic areas and no improvement with folic acid and antibiotics |
| Intestinal lymphoma | Normal β2 microglobulin; Exclusion on intraoperative enteroscopy and jejunal biopsies |
| Adult-onset autoimmune enteropathya | Significative intraepithelial lymphocytosis is against this diagnosis (it would be expected reduced number of IEL); positive HLA DQ2/DQ8 is not typical of this condition |
| Hypogammaglobulinemia | Normal serum immunoglobulin and protein electrophoresis; no major changes in thoracic CT scan; no history of recurrent infections |
| Small-bowel ischaemia | Histology not compatible |
| Drug-associated enteropathy | No history of suspicious medication |
 CD3 positive, but (B) mostly CD8 negative and (C) CD30 negative.")


Celiac disease is a common autoimmune condition that is generally associated with good prognosis as the compliance of a strict GFD leads to clinical and mucosal recovery.1 When diagnosed in adults, particularly above the age of 50, the presentation is often atypical and lack of improvement under GFD is more likely.4 Iron deficiency and extraintestinal manifestations (like bone disease and depression) can be the only presenting signs of the disease, and definitive diagnosis is normally delayed.9 At this stage, the diagnostic workup is significantly more complex, especially in cases of seronegative CD. In our particular case, although the clinical suspicion and the first duodenal biopsies (taken while the patient was on normal diet) corroborated the diagnosis of CD, there were two major problems – negative celiac-specific antibodies performed while on a gluten-containing diet and unresponsiveness to GFD. In cases of non-responsive CD (NRCD), after ensuring strict adherence to GFD, the first step is to confirm the initial diagnosis of CD and exclude other entities that mimic CD symptoms and other causes of villous atrophy. The positivity of celiac-specific antibodies is one of the parameters used to confirm the diagnosis, but serology may be negative in 5–16% of patients with biopsy-confirmed CD.2,3,5 This was the case of our patient that despite negative specific serology presented typical histology and high clinical suspicion. In these ambiguous seronegative CD cases HLA typing may help to rule out or confirm the diagnosis, as HLA-DQ2 (≈95%) or HLA-DQ8 (≈5%) are present in almost all patients with CD.10,11 So, CD is excluded for patients who are negative for HLA-DQ2 or HLA-DQ8, considering its high negative predictive value (>99%).12,13 In fact, our patient presented HLA DQ2/DQ8 compound heterozygosity, supporting the CD diagnosis.
NRCD is common, affecting from 7% to 30% of patients under GFD. There are many distinct aetiologies including inadvertent gluten ingestion (the most common cause), other food intolerances, small intestinal bacterial overgrowth, microscopic colitis, pancreatic insufficiency and RCD, that makes up a small subset (approximately 10%) of NRCD.14–16 In fact, during the extensive investigation to exclude other potential causes of chronic diarrhoea (Table 1), we identified two concomitant entities – microscopic colitis and giardiasis – that could be responsible for some persisting symptoms, but despite being successfully treated, the severe symptoms persisted. Moreover, these are two common conditions associated with CD, particularly microscopic colitis that develops in 4% of patients with CD.9
RCD is almost exclusive of patients diagnosed with CD above the age of 50, and two to three times more frequent in women.7 Our patient had all these epidemiological features. The immunohistochemical studies as well as a T-cell receptor gene rearrangement analysis showing clonal expansion were indicative of RCD-II.7 This diagnosis was further supported by the presence of severe and life threatening malnutrition, protein losing enteropathy, bowel-wall thickening in the jejunum/ileum and mesenteric lymphadenopathy at CT scan enteroclysis and, most important, evidence of ulcerative jejunoileitis, that should be considered synonym of RCD-II.8,17,18 When there are doubts about the diagnosis in immunohistochemical and/or T-cell receptor gene rearrangement, flow cytometry is a good option, since it is able to differentiate cytoplasmatic from membranous CD3 expression and to identify other aberrant IEL with preserved CD8 expression.7 EATL is a threatening complication and was a major concern in this patient, considering the clinical severity and lack of sustained response to medical treatment. However, its’ presence was systematically excluded in multiple histological analyses. Additionally, the aberrant IELs of our patient lack CD30 expression that is associated with worse prognosis, including the development of overt EATL.19
RCD-II has a poor prognosis, with a five-year survival between 44 and 58%. The higher mortality can be largely attributed to the high risk of developing EATL, which occurs between 33% and 52% within 5 years after diagnosis.7 Even in the absence of EATL RCD-II is often complicated with threatening haemorrhage due to ulcerative jejunoileitis, severe malabsortion with intestinal failure and severe sepsis, as occurred in our patient.5,9
As RCD-II is a rare disease, to date there is no standardised therapy, and the evidence for treatment is based on case reports, open-label observational or prospective experiences, and expert opinion.17 Apart from TPN and substitution of vitamins, that are consensual, pharmacological therapy is question of debate. Immunosuppressive drugs suggested for RCD-I include steroids, thiopurines, cyclosporine, infliximab and alemtuzumab.8,17 Steroids suppress clinical symptoms in RCD-I and clinical improvement is reported in up to 90% of patients.8 However, RCD-II is, at least in part, resistant to most therapies evaluated so far and there is no place for immunosuppressive drugs such as azathioprine and infliximab, that could increase the risk of progression to EATL.4,8,17 Cladribine is equally toxic to proliferating as to nondividing lymphoid cells, and this feature is particularly interesting for the treatment of low-grade malignancies, as RCD-II is considered, and there are already some published cases with favourable results.7,20 We decided to use this strategy in our patient but it did not lead to sustained improvement. Finally, high-dose chemotherapy followed by ASCT has been explored for RCD-II in a pilot study from a single centre.21 This was our last alternative, but unfortunately it did not bring sustained clinical or histological improvements. Interleukin-15 blockade could also be considered as an experimental therapy.22
Surgery in RCD should be limited to the management of complications such as perforation, massive haemorrhage, high-grade obstruction, and cancer.8,23
The present case also emphasises the uncertainties regarding the optimal follow-up strategy of patients with RCD-II. Regular follow-up, including UGI endoscopy with biopsies, CT scan or MR enteroclysis, and PET scan, is necessary to detect EATL as early as possible.7 However, the best tests for EATL surveillance and the appropriate interval between examinations are not currently established.8 Clinical supervision in RCD should not be limited to EATL surveillance but should also include monitoring for nutritional deficiencies and bone density measurements.
This RCD-II case demonstrates the difficulties in the diagnosis, management and surveillance of this rare condition. The therapeutic approach is complex, since there are few options, whose efficacy is not yet fully demonstrated, and which are not sufficiently validated.
In our opinion, due to rarity of RCD, these patients should be referred to specialised centres. International consensuses in these areas are urgently required in order to facilitate future therapeutic advances.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that the procedures followed were in accordance with the regulations of the relevant clinical research ethics committee and with those of the Code of Ethics of the World Medical Association (Declaration of Helsinki).
Confidentiality of dataThe authors declare that they have followed the protocols of their work centre on the publication of patient data.
Right to privacy and informed consentThe authors declare that no patient data appear in this article.
Conflicts of interestThe authors have no conflicts of interest to declare.
Chris Mulder, MD, PhD (Department of Gastroenterology and Hepatology, Celiac Centre Amsterdam, VU University Medical Centre, Amsterdam, The Netherlands) for the complementary histological analysis and therapeutic guidance.
