


The LILRB1 gene has recently been associated with rheumatoid arthritis (RA) susceptibility in HLA-DRB1-shared epitope (SE) negative Japanese individuals. Since the contribution of the LILRB1 polymorphism to RA susceptibility may vary among ethnic populations, we examined this association in a group of Caucasian patients. The frequency of LILRB1 alleles was also determined in patients according to the presence of DRB1-SE.
MethodsSamples from 103 RA patients and 107 healthy controls were randomly collected. Polymorphism of the LILRB1 gene was analyzed by sequencing with primers that amplified intron 3 and exon 4.
ResultsThe frequencies of LILRB1 alleles in RA patients did not differ from those of controls. However, when patients and controls were grouped according to SE, the PE-01/01 genotype was less frequent in negative-SE patients than in controls. Whereas SE is associated with higher anti-CCP antibody levels, as expected, the production of anti-CCP antibodies was lower in negative-SE patients with PE-01/01 genotype. Moreover, radiographic damage in hand and feet of SE-negative PE-01/01 patients was less severe than in patients with other genotypes.
ConclusionsThe participation of this LILRB1 polymorphism in the RA pathogenesis of this Caucasian cohort differed from that reported in a Japanese sample. Our findings suggest that the LILRB1-PE-01/01 genotype could exert a protective role in RA susceptibility and disease severity in the absence of SE.
El gen LILRB1 se ha asociado a la susceptibilidad a desarrollar artritis reumatoide (AR) en aquellos individuos japoneses negativos para el epítopo compartido del HLA-DRB1. Dado que la contribución del polimorfismo en LILRB1 a la susceptibilidad a desarrollar artritis reumatoide puede variar según las distintas etnias, hemos examinado esta asociación con la artritis reumatoide en un grupo de pacientes caucásicos. La frecuencia de los alelos LILRB1 fue también determinada en los pacientes clasificados según la presencia del epítopo compartido.
MétodosSe recogieron al azar muestras de 103 pacientes de AR y 107 controles sanos. El polimorfismo en el gen LILRB1 se analizó con cebadores que amplificaban el intrón 3 y el exón 4.
ResultadosLas frecuencias diploides de los alelos de LILRB1 en los pacientes de artritis reumatoide no diferían de la de los controles. Sin embargo, cuando los pacientes y controles se agruparon según la presencia del epítopo compartido, la frecuencia del genotipo PE-01/01 fue menor en los pacientes con epítopo compartido negativo que en controles con epítopo compartido negativo. Mientras que el epítopo compartido se asoció con niveles altos de anticuerpos anti-CCP, la producción de anticuerpos anti-CCP fue menor en pacientes con epítopo compartido negativo y genotipo PE-01/01. Además, el daño radiográfico en manos y pies en pacientes con epítopo compartido negativo y genotipo PE-01/01 fue menos severa que en pacientes con otros genotipos.
ConclusionesLa participación del polimorfismo del LILRB1 en la fisiopatogenia de la AR en esta cohorte de pacientes Caucásicos difiere de la publicada en los pacientes japoneses. Nuestros resultados sugieren que el genotipo LILRB1-PE-01/01 puede ejercer un papel en la susceptibilidad de la AR y en la severidad de la misma en ausencia del epítopo compartido.
Rheumatoid arthritis (RA) is a systemic inflammatory disorder characterized by chronic inflammation and destruction of the synovial joints leading to progressive joint damage and disability. It is a clinically heterogeneous condition with a complex etiology in which environmental and genetic factors are implicated.
Data from familial and twin studies suggest that 60% of RA susceptibility is due to genetic factors.1 Prevalence ranges from 2 to 12% in first-degree relatives of patients, whereas the population prevalence is only 0.2 to 1%.2 Concordance rates range from 12% to 30% in monozygotic twins and from 5% to 10% in same-sex dizygotic twins.3,4
Human leukocyte antigens (HLA) were the first genes to be associated with RA susceptibility. HLA-DRB1 alleles involved in this disease are characterized by the presence of a common amino acid sequence (QKRAA, QRRAA, RRRAA). This shared epitope (SE) is located in the third hypervariable region of certain HLA-DRB1 molecules.5–7 Additional candidate gene polymorphisms have been linked to RA: protein tyrosine phosphatase non-receptor type 22 (PTPN22), involved in HLA class II dependent T cell activation8; cytotoxic T lymphocyte antigen-4 (CTLA-4), an important negative regulator of T cell activation8; peptydil arginil deiminase isoform 4 (PADI 4), an enzyme involved in the post-translational deimination of protein bound arginine to citrulline8; and tumor necrosis factor receptor II (TNFRII), capable of inducing the nuclear factor κB and apoptosis.9
A recent report based on a sample of Japanese HLA-DRB1 negative patients has associated an allele of the leukocyte immunoglobulin-like receptor subfamily B member 1 gene (LILRB1) with RA.10 LILRB1 belongs to a family of inhibitory and stimulatory receptors located in the leukocyte receptor complex on human chromosome 19q13.4.11 It has a broad cellular distribution including cells of both the myeloid and lymphoid origin: peripheral blood monocytes, in vitro-derived dendritic cells and macrophages, B cells, and a subset of T and NK cells.12–14 Its binding site interacts with the well conserved α3 domain of HLA class I competing directly with CD8 for binding to MHC-I,15 and the analogous region in the Cytomegalovirus (CMV) UL18 protein cells.12–14 Its cytoplasmic domain delivers inhibitory signal through immunoreceptor tyrosine-based inhibition motifs (ITIMs).16 The blocking of CD8 binding to MHC and the presence of ITIMs suggests that LILRB1 has a crucial role in the modulation of immune responses.
The contribution of the LILRB1 polymorphism to RA susceptibility probably differs among ethnic populations. We, therefore, examined the association of LILRB1 polymorphisms with RA in a sample of Caucasian patients. Due to the substantial contribution of the HLA-DRB1-shared epitope in RA susceptibility and the ethnic variations in the association of RA with certain DR alleles, we also performed a stratified analysis of the frequency of LILRB1 genotypes according to the presence of a shared epitope in these patients. The influence of LILRB1 polymorphism on autoantibody production and clinical activity was analyzed.
Patients and methodsPatientsSamples from 103 RA patients were recruited from the Rheumatology Department at Hospital de la Santa Creu i Sant Pau, Barcelona, Spain. All experiments received prior approval from the Hospital de Sant Pau Institutional Ethics Committees. Patients fulfilled the American College of Rheumatology (ACR; formerly the American Rheumatism Association) 1987 revised criteria for RA.17 They were stratified according to HLA-DR in order to assess the association of the polymorphism in the presence or absence of SE with the disease phenotype: levels of rheumatic factor (RF), cyclic citrullinated peptide antibodies (CCP) and Sharp-van der Heijde score. Among the RA patients, 70.8% were females, mean±standard deviation (SD) age was 59.6±12.2, age at disease onset was 46.81±14.43, the evolution period was 13.29±9.15 and 52.43% were positive for SE. Samples from anonymous 107 healthy controls were also randomly recruited from Hospital de la Santa Creu I Sant Pau.
In all subjects, genomic DNA was extracted from blood leukocytes, using QIAamp® DNA Blood Mini Kit (Qiagen, Germantown, MD).
LILRB1 intron 3 and exon 4 single nucleotide polymorphism (SNP) genotypingPolymorphism screening was performed by polymerase chain reaction (PCR)-sequence based typing. The primers were designed according to the genomic DNA sequence of human LILRB1 (GenBank accession N. AF189277). The set of primers used was: 5′-TGAGTCTGTCCCCAGCTCTT-3′ in position c.71–192 at intron 3 (forward: F) and 5′-AGGGGGTCACTGCTCTCTG-3′ in position c.342 at exon 4 (reverse: R). PCR was performed in 50μL reaction mix containing 100–200ng of genomic DNA. Thermal cycling program was: initial denaturation: 95°C for 3min; 34 cycles of denaturation, annealing and extension consisting in: denaturation: 95°C for 45s; annealing: 55.4°C for 30s; extension: 72°C for 1min; last extension: 72°C for 5min. ExoSAP-IT was used to cleanup unconsumed deoxyribonucleotide triphosphate and primers (USB Corporation, Ohio, USA). Sequencing reaction was then performed using the R primer and the ABI PRISM BigDye terminator v1.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA). Thermal cycling program was: initial denaturation: 96°C for 10s; 34 cycles of denaturation, annealing and extension consisting in: denaturation: 96°C for 10s; annealing: 50°C for 10s; extension: 60°C for 2min. After amplification, the excess of dye-labeled dideoxynucleotides was removed using AutoSeqTMG-50 columns (GE Healthcare UK Limited, Amersham, UK). The purified products were analyzed with an ABI Prism 3100 (Applied Biosystem).
HLA-DRB1 genotypingHLA-DRB1 low resolution typing was performed by PCR-sequence specific oligonucleotide (SSO). DNA samples were amplified using a master mix containing five biotinylated sequence-specific primers (Dynal RELI SSO HLA-DRB, Dynal Biotech GmbH, Hamburg, Germany) as previously described.18 HLA-DRB1 high resolution typing of DRB1*01 DRB1*04 and DRB1*14 (Dynal AllSet+™, Dynal Biotech GmbH) was performed by PCR-single sequence polymorphism (SSP) as previously described.18 The SE was defined by the following alleles: DRB1*0101, *0102, *0401, *0404, *0405, *0408, *0413, *1001 and *1402.19,20
Measure of serological markers of RAAnti-CCP antibodies (Abs) were detected by EliA™ on ImmunoCAP® 250 Phadia (Freiburg, Germany), considering values above 10U/mL as positive. RF was detected by nephelometry with Beckman Coulter IMMAGE® Immunochemistry System (Fullerton, USA), values above 20 International Units (IU)/mL were considered positive. All these measures were detected at the same period of time.
Radiographic analysisRadiographs were analyzed by the Sharp-van der Heijde method.21 In brief, erosion was assessed in 16 joints for each hand and wrist, and six joints for each foot. One point was scored if erosions are discrete, rising to 2, 3, 4, or 5 depending on the amount of surface area affected; the score for erosion ranged from 0 to 160 in the hands and from 0 to 120 in the feet. Joint space narrowing (JSN) was assessed in 15 joints for each hand and wrist, and six joints for each foot. JSN was scored 0 for normal, 1 for focal or doubtful, 2 for generalized less than 50%, 3 for generalized more than 50% and 4 for bony ankylosis or complete luxation.
Statistical analysisGenotype frequencies of the patients and controls were compared by the χ2 test or Fisher exact test. Odds ratio (OR) and confidence intervals (CI) were calculated to estimate relative risks of each genotype. Serological and clinical markers were compared by the t-test. For all tests, p values less than 0.05 were considered statistically significant. Calculations were performed using the SPSS V 15.0 package (SPSS Inc., Chicago, IL, USA).
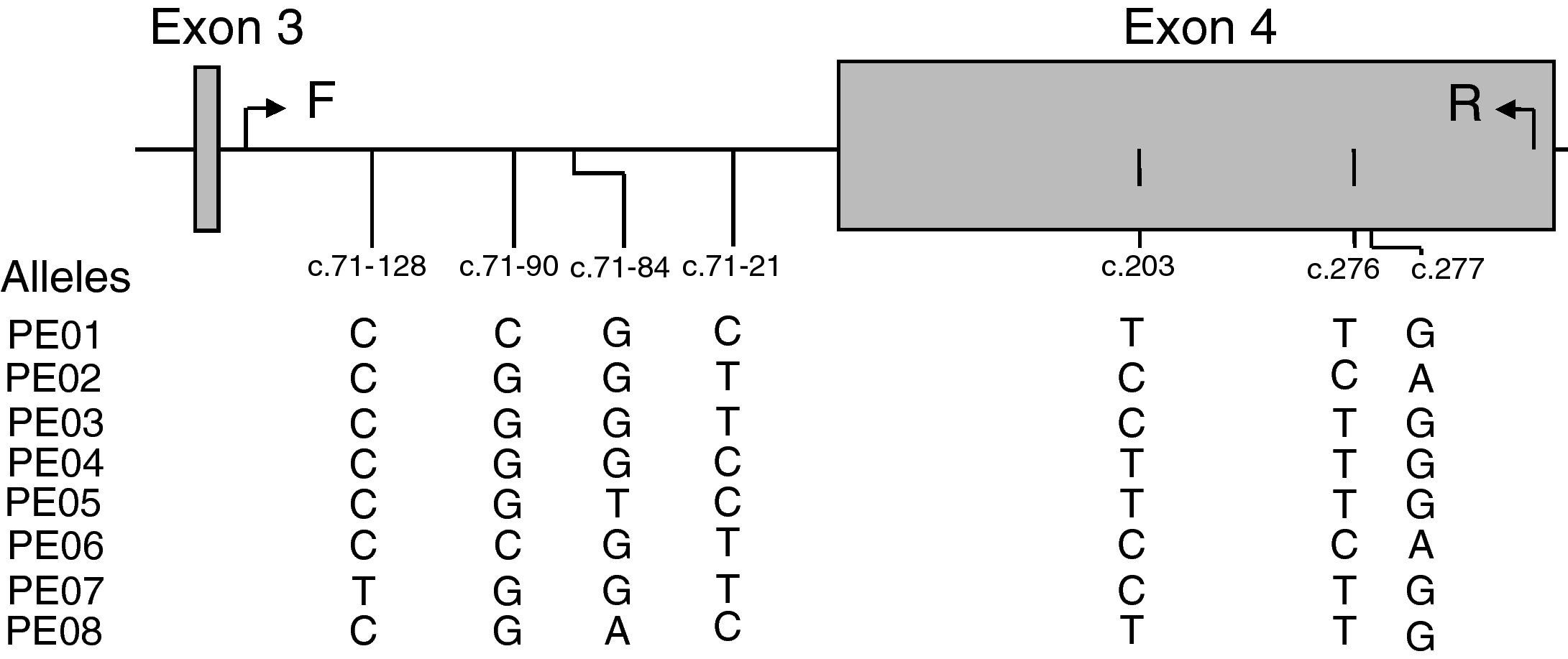
ResultsAnalysis of SNPs in the extracellular region (exon 4) of LILRB1 in a Caucasian populationEight different SNPs were detected using primers that amplified a 497bp fragment from position c.71–192 at intron 3 to c.342 at exon 4 (Fig. 1). Five of these SNPs were in non-coding regions at positions c.71–128 C>T (new SNP), c.71–90 C>G, c.71–84 G>T>A and c.71–21 C>T, and 3 SNPs were in coding regions at position c.203 T>C (P68L, rs1061679), c.276 T>C (H96H), and c.277 G>A (A93T, rs12460501). These 8 SNPs were named according to Kuroki's nomenclature (PE-01 to PE-08) to facilitate comparison with Japanese population results.10 However, it should be mentioned that the LILRB1 fragment analyzed in the present study included exon 4 and partial intron 3.
Association of LILRB1 polymorphism with RA susceptibility.")
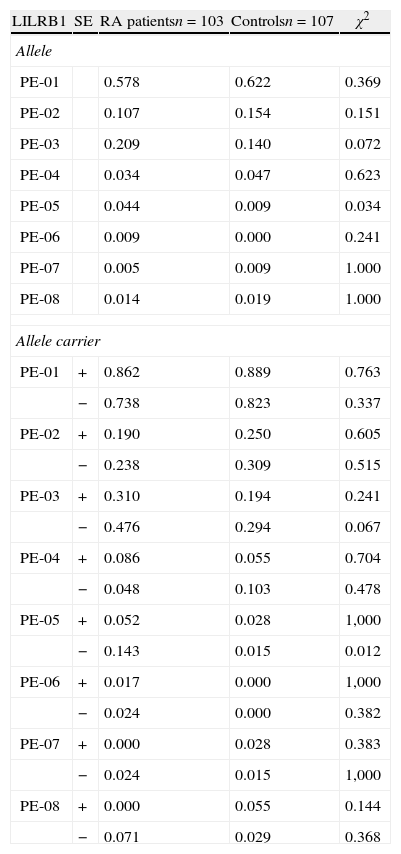
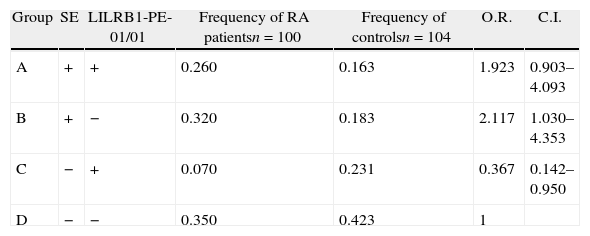
The LILRB1 polymorphism in the extracellular region was determined in 103 RA patients and in 107 healthy controls, as shown in Table 1. The allele frequencies of RA patients and controls were not significantly different except for allele PE-05, which was more frequent in RA patients than controls (p=0.034). The genotype and allele distribution of PE-01, PE-02 and PE-03 polymorphisms were in agreement with the Hardy–Weinberg equilibrium among the patients and control group. We then analyzed the allele carrier frequency in healthy controls and patients segregated by the presence of HLA-DRB1 SE. A significant association of PE-05 was detected in HLA-DRB1 SE negative RA patients (p=0.012, OR=11.167, CI: 1.294–96.383). Genotype frequencies were also evaluated in patients and healthy controls with or without HLA-DRB1 SE presence. Only the genotype PE-01/01 was significantly increased in controls in the absence of SE (p=0.04). None of the other genotypes had statistically significant differences between patients and controls with or without SE. Since the frequency of the allele PE-05 was low in this cohort, neither patients nor controls with the PE-05/05 genotype were found. Table 2 shows patients and controls classified in four groups according to the LILRB1-PE-01/01 genotype and the presence of SE. No difference was observed in the frequency of PE-01/01 patients and healthy controls with SE (p=0.835). However, a significant increase was observed in the frequency of PE-01/01 healthy controls without SE when compared with SE negative patients (OR 0.367, CI=0.142–0.950). The genotype PE01/01 was thus underrepresented in SE negative patients.
Frequencies of LILRB1 alleles in Caucasian population.
| LILRB1 | SE | RA patientsn=103 | Controlsn=107 | χ2 |
| Allele | ||||
| PE-01 | 0.578 | 0.622 | 0.369 | |
| PE-02 | 0.107 | 0.154 | 0.151 | |
| PE-03 | 0.209 | 0.140 | 0.072 | |
| PE-04 | 0.034 | 0.047 | 0.623 | |
| PE-05 | 0.044 | 0.009 | 0.034 | |
| PE-06 | 0.009 | 0.000 | 0.241 | |
| PE-07 | 0.005 | 0.009 | 1.000 | |
| PE-08 | 0.014 | 0.019 | 1.000 | |
| Allele carrier | ||||
| PE-01 | + | 0.862 | 0.889 | 0.763 |
| − | 0.738 | 0.823 | 0.337 | |
| PE-02 | + | 0.190 | 0.250 | 0.605 |
| − | 0.238 | 0.309 | 0.515 | |
| PE-03 | + | 0.310 | 0.194 | 0.241 |
| − | 0.476 | 0.294 | 0.067 | |
| PE-04 | + | 0.086 | 0.055 | 0.704 |
| − | 0.048 | 0.103 | 0.478 | |
| PE-05 | + | 0.052 | 0.028 | 1,000 |
| − | 0.143 | 0.015 | 0.012 | |
| PE-06 | + | 0.017 | 0.000 | 1,000 |
| − | 0.024 | 0.000 | 0.382 | |
| PE-07 | + | 0.000 | 0.028 | 0.383 |
| − | 0.024 | 0.015 | 1,000 | |
| PE-08 | + | 0.000 | 0.055 | 0.144 |
| − | 0.071 | 0.029 | 0.368 | |
SE=shared epitope.
Association of LILRB1-PE-01/01 and SE with RA susceptibility.
| Group | SE | LILRB1-PE-01/01 | Frequency of RA patientsn=100 | Frequency of controlsn=104 | O.R. | C.I. |
| A | + | + | 0.260 | 0.163 | 1.923 | 0.903–4.093 |
| B | + | − | 0.320 | 0.183 | 2.117 | 1.030–4.353 |
| C | − | + | 0.070 | 0.231 | 0.367 | 0.142–0.950 |
| D | − | − | 0.350 | 0.423 | 1 |
A vs. B, p=0.835; A vs. D, p=0.129; B vs. D, p=0.049; C vs. D, p=0.049.
Odds ratio (O.R.) was calculated against individuals in group D. SE=shared epitope.
Statistical test: two-sided t-test.
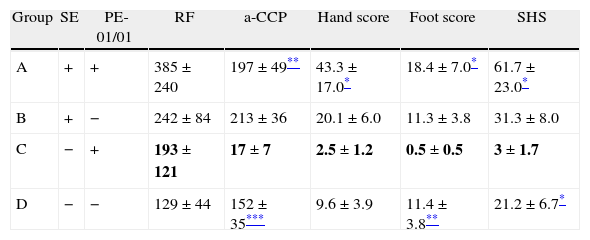
A detailed review of the clinical history of patients carrying PE-05 allele did not reveal any distinguishing trait in these subjects. We then examined the association between LILRB1 PE-01/01 genotype and the presence of RF or anti-CCP antibodies. The titer of anti-CCP antibodies was expectedly higher in SE positive than in SE negative patients (199±18 vs. 122±21 respectively, p=0.008). Patients were then segregated according to the presence of SE and PE-01/01 genotype to evaluate the association of these autoantibodies with LILRB1 polymorphisms. The titer of RF and anti-CCP antibodies during the first 10 years of disease was evaluated in the four groups established according to SE and PE-01/01 (Table 3). No significant difference in the RF titer was observed in any of the patient groups. The presence of SE significantly increased the anti-CCP antibody titer in PE-01/01 patients (197±49 vs. 17±7, p=0.007). On the other hand, the PE-01/01 genotype was associated with a lower titer anti-CCP antibodies in SE negative patients (17±7 vs. 152±35, p=0.002). None of the other LILRB1 genotypes were associated with anti-CCP or RF titer in SE positive or SE negative patients (data not shown).
Association of LILRB1 polymorphisms and SE with RA severity.
| Group | SE | PE-01/01 | RF | a-CCP | Hand score | Foot score | SHS |
| A | + | + | 385±240 | 197±49** | 43.3±17.0* | 18.4±7.0* | 61.7±23.0* |
| B | + | − | 242±84 | 213±36 | 20.1±6.0 | 11.3±3.8 | 31.3±8.0 |
| C | − | + | 193±121 | 17±7 | 2.5±1.2 | 0.5±0.5 | 3±1.7 |
| D | − | − | 129±44 | 152±35*** | 9.6±3.9 | 11.4±3.8** | 21.2±6.7* |
All comparisons were established with C group (SE negative and PE-01/01 positive patients, in bold).
SE=shared epitope. Statistical test: two-sided t-test.
To evaluate the association of LILRB1 polymorphism with clinical activity, radiological damage in the first 10 years of follow-up was measured using the Sharp van der Heijde score (SHS). Patients were then segregated into four groups according to their LILRB1 genotype and SE presence. The erosion and joint space narrowing scores of hands and feet and the total SHS of each group of patients are shown in Table 3. There was a significant difference in the hand score and total SHS of SE positive and SE negative patients (hand score: 49.6±9.1 vs. 23.4±4.4, p=0.012, total score: 69.2±12.9 vs. 38.1±8, p=0.04).
The presence of SE significantly increased radiographic damage in hands and feet and consequently in the SHS of PE-01/01 positive patients (hand: 43.3±17 vs. 2.5±1.2, p=0.04; feet: 18.4±7 vs. 0.5±0.5, p=0.04; and SHS: 61.7±23 vs. 3±1.7, p=0.03). In the absence of SE, SHS was significantly lower in patients with PE-01/01 genotype (3±1.7 vs. 21.2±6.7, p=0.02). When analyzing hand and foot damage separately, the genotype PE-01/01 was associated with significantly fewer erosions and joint space narrowings in SE negative patients (feet: 0.5±0.5 vs. 11.4±3.8, p=0.013; hand: 2.5±1.2 vs. 9.6±3.9, p=0.09).
DiscussionFindings in this Caucasian cohort from Southwestern Europe suggest a negative association between the LILRB1-PE-01/01 genotype and RA. Similarly, to Japanese RA patients, the effect of the PE-01/01 genotype on RA risk only became apparent after excluding the influence of SE. The LILRB1-PE-01/01 genotype was positively associated with RA in Japanese patients.10 However, in our cohort the frequency of LILRB1-PE-01/01 genotype was significantly lower in patients lacking SE when compared with controls. Since the genotyping methods used in both studies were identical, the discrepancy could be attributable to different characteristics of the cohorts or variations between the two ethnic groups. Susceptibility to some diseases varies with race and ethnicity and linkage of complex diseases in a specific population is not consistently replicated in other racial and ethnic groups.22 A number of genes have shown population-specific associations with RA susceptibility, but results with the same genes in other populations have been contradictory.23,24 Additional replications with case-control cohorts in other populations would confirm the LILRB1 association to RA.
This discrepancy between our results and those of Kuroki's report could be the consequence of the different genotype and allele frequencies of LILRB1 in Japanese and Caucasian populations. There may also be a differential LILRB1–HLA interaction depending on the SE allele. Indeed, not all SE genotypes confer the same relative risk of RA25,26 and each population has a characteristic distribution of SE genotypes. Accordingly, the most common SE encoding allele in our population is HLA-DRB1*0101 (the genotype frequency in the population is 0.057 and in RA patients it is 0.134)27, whereas in the Japanese population it is HLA-DRB1*0405 (the genotype frequency is 0.139 in the control population and 0.295 in RA patients).28
Our results suggest that anti-CCP-positive or -negative disease can be associated with distinct genetic risk factors. The significant association of HLA-DRB1 SE+ genotype with higher anti-CCP titers in PE-01/01 patients is consistent with the previously reported clear gene-dose effect of SE on anti-CCP antibodies.29 Since patients with anti-CCP antibodies have more swollen joints and more severe radiological destruction than patients without such antibodies, the linkage of SE with radiographic severity may be indirectly due to SE association with anti-CCP. If this were so, it would not be surprising that LILRB1 PE-01/01 could be associated with lower levels of anti-CCP antibodies and less severe radiographic outcome. It is possible that anti-CCP positive RA disease is a separate pathogenic entity associated with SE and LILRB1. Van der Helm et al. have reported that the disease course of RA patients with anti-CCP antibodies differed from that of patients without these antibodies.30 Furthermore, SE and LILRB1 loci could be associated with different RA traits, and certain alleles could confer particular characteristics for the disease. Otherwise, the ligation of LILRB1 on dendritic cells by self-HLA molecules may play a role in controlling the balance between the induction and suppression of adaptive immune responses31 Since RA is genetically heterogeneous, gene-based stratifications based on the SE and LILRB1 could be useful to identify clinical subsets of patients. SE-negative and LILRB1-PE-01/01 patients would, therefore, be characterized by a lower level of antibodies and disease activity. Although these findings cannot be generalized to all populations, management could be less aggressive in this group of patients. Incorporation of this gene-based stratification could also facilitate the genetic and molecular dissection of RA disease. The frequent association of RA with other autoimmune diseases could be dissected by analogous gene-based analysis.
The association of LILRB1 alleles with RA suggests that this molecule could play a role in the etiopathological mechanisms of the disease. LILRB1 molecules are preferentially present in active inflammatory conditions characterized by extensive leukocyte infiltration, LILRB1 may act as an inhibitory feedback in psoriatic skin32 and in multiple sclerosis brain.33 Moreover, the LILRB1 gene family is located in 19q13.4, one of the candidate susceptibility regions for systemic lupus erythematosus and inflammatory or infectious diseases such as spondylarthropathies and leprosy.34,35 These reports suggest that LILRB1 may have a role in the maintenance of immunological tolerance. The broad recognition of MHC-I molecules by LILRB1 inhibits NK cell activity and T cell cytotoxicity.13,36 Although LILRB1 binds to conserved domains in MHC alleles, LILRB1 protein changes coded by different alleles might affect protein-protein interactions with functional consequences. On the other hand, LILRB1 has a high affinity for the human CMV MHC-I homologous gpUL18. The human cytomegalovirus MHC-I homologous UL18 inhibits LILRB1 positive cells through a direct CMV UL18-LILRB1 interaction.37 An alteration in the affinity of LILRB1 for human CMV UL18 or other unidentified ligands could be involved in the susceptibility to inflammation. Despite the association found, further investigation is required to demonstrate the role of LILRB1 as a regulator of inflammation. LILRB1 could then be considered a novel therapeutic target in pathological inflammatory processes.
Conflict of interestThere is no conflict of interest.
We thank Carolyn Newey for editorial assistance and Ignasi J. Gich for advice in the statistical analysis.
E.C. is a participant in the Program “Contratos de apoyo a la Investigacion del Sistema Nacional de Salud”. S.V. was supported by “Fondo Investigaciones Sanitarias” and participant in the Program for Stabilization of Investigators of the “Direccio d’Estrategia i Coordinacio del Departament Salut de la Generalitat de Catalunya”.