


Este estudio fue diseñado para examinar el efecto del HGF sobre la inflamación vascular e hipertensión en ratas SHR. Nosotros especulamos que la disminución vascular del HGF puede desempeñar un papel fundamental en la patogénesis de la hipertensión arterial en SHR, y que el incremento de los niveles de HGF puede producir una disminución en la presión arterial a través de la reducción de la inflamación vascular.
Materiales y métodosSe utilizaron SHR de 14 semanas de edad, a las cuales se les administró el gen del HGF humano (1mg/kg) por vía hidrodinámica (SHR-pCMV-HGF, n=6) o el vector vacío (SHR-pcDNA3.1, n=6) por 6 semanas. Los controles fueron WKY (n=6). La presión arterial sistólica fue medida semanalmente. La activación del factor NF-kB fue evaluada en la fracción nuclear mediante Western blot, la expresión de mediadores proinflamatorios mediante RT-qPCR y Western blot.
ResultadosLa presión arterial, la activación del NF-κB y la expresión de IL-6, MCP-1 y RANTES fueron significativamente más elevadas en SHR que en WKY. La terapia génica con el HGF normalizó la actividad vascular del NF-κB, suprimió la expresión de los mediadores inflamatorios y redujo la HTA.
ConclusiónEste estudio sugiere que la disminución en la concentración de HGF en la aorta ejerce un papel importante en la activación de mediadores proinflamatorios observados en SHR y sugiere que el HGF puede representar un potencial agente terapéutico en el tratamiento de la HTA.
This study was designed to examine the effects of hepatocyte growth factor (HGF) gene delivery on vascular inflammation and hypertension in spontaneously hypertensive rats (SHR). We speculated that HGF deficiency could play a key role in the pathogenesis of hypertension in SHR, and that increasing HGF levels will produce prolonged decreases in blood pressure due to reduced vascular inflammation.
Materials and methodsFifteen-week old male SHRs received weekly hydrodynamic injections of a naked plasmid containing human HGF (pCMV-HGF) (1mg/kg) or empty vector (pcDNA3.1) for 6 weeks. Two groups of Wistar-Kyoto (WKY) rats were used as controls (n=6) and treated in the same manner. The activation of NF-κB was assessed by Western blot and mRNA expression of pro-inflammatory cytokines by real-time PCR and Western blot.
ResultsBlood pressure, NF-κB activation and expression of IL-6, MCP-1 and RANTES were significantly higher in SHR than in the control WKY. The HGF gene therapy normalized NF-κB activity, pro-inflammatory cytokines expression, and decreased the hypertension in SHR.
ConclusionThese observations suggest that decreased aorta HGF concentration may have a role in the vascular inflammation observed in SHR, and demonstrate that increasing HGF is a potential therapeutic target in the treatment of hypertension.
La inflamación es un proceso coordinado por secreción local de moléculas de adhesión, factores quimiotácticos y citocinas, cuya expresión es el resultado del daño vascular1. Evidencias experimentales sugieren que mediadores proinflamatorios a nivel vascular participan de manera importante en la fisiopatología de la hipertensión arterial (HTA)2. Otros efectores vasculares, como el sistema renina-angiotensina-aldosterona, parecen contribuir con los mediadores inflamatorios en la etiopatogenia de la HTA3, induciendo la remodelación de la pared del vaso y el desarrollo de ateroesclerosis4,5.
La angiotensina ii (AII) también es considerada un mediador proinflamatorio que tiene un rol importante en el desarrollo de mecanismos inflamatorios relacionados con complicaciones en la ateroesclerosis. EL papel de la AII involucra la activación de factores de transcripción como el factor nuclear kB (NF-kB)4, el cual es capaz de aumentar la expresión de numerosos mediadores proinflamatorios como citocinas, quimiocinas y moléculas de adhesión4.
Estudios previos han demostrado que la administración de inhibidores de la enzima convertidora de angiotensina o antagonistas de los receptores tipo 1 de la AII (AT1) disminuye el daño endotelial y la remodelación vascular en hipertensión clínica y experimental6.
De igual forma, se ha asociado el incremento en los niveles de RANTES, un potente agente quimiotáctico miembro de la familia de las quimiocinas CC7, al proceso inflamatorio observado en la hipertensión inducida por AII8, así como también se han reportado niveles elevados de MCP-19. De igual forma, un estudio reciente demostró un incremento significativo en la expresión de las citocinas IL-6, IL-1β y TNF-α y de las moléculas de adhesión ICAM-1 y VCAM en la pared vascular de SHR10.
Un evento fundamental en la fisiopatología de la inflamación es la activación del factor de transcripción NF-kB, el cual controla la expresión de numerosas moléculas proinflamatorias11. Diversos estudios han demostrado que HGF suprime la activación del NF-kB inducida por TNF-α en células cultivadas12 y está implicado en la regeneración de la célula endotelial capilar13.
El HGF es una citocina pleiotrópica y multifuncional que activa múltiples vías de señalización, a través de las cuales modula una variedad de procesos celulares como efectos mitogénicos y antiapoptóticos14. El HGF se produce en forma inactiva como una cadena única por fibroblastos, células del mesénquima, células endoteliales, células del hígado15 y es esencial en la regulación de la proliferación, en la diferenciación y en la supervivencia celular en diversos órganos16. In vivo el HGF protege contra el daño agudo y crónico en el hígado17, el intestino9 y el riñón16, y se ha sugerido como mecanismo mediador de esta acción su efecto antiinflamatorio12.
El HGF inhibe la infiltración de neutrófilos disminuyendo la regulación de moléculas de adhesión, tales como ICAM-1/E-selectina sobre la superficie de la célula endotelial15. Asimismo, esta proteína suprime el daño isquémico18.
Por otra parte, el HGF posee un marcado potencial antifibrótico, ya que inhibe la producción de citocinas fibrogénicas como el TGF-β1 en tejido cardíaco y miofibroblastos de hámster cultivados19, y también disminuye la expresión de TGF-β1 y su receptor en el epitelio tubular en un modelo de fibrosis renal en ratón20,21; además, la acción antifibrótica del HGF exógeno estuvo acompañada por una menor inflamación renal en un modelo de enfermedad renal crónica12.
Estudios previos demostraron que la expresión de HGF se encuentra disminuida en la aorta de SHR, sin embargo, se desconoce si esta reducción en los niveles locales de HGF desempeña un papel importante en el desarrollo de la HTA en SHR. De esta manera, nosotros evaluamos el efecto de la sobreexpresión del HGF en el desarrollo de hipertensión arterial en SHR. Nuestros resultados demuestran que la administración de HGF exógeno produce una disminución significativa de la presión arterial en este modelo experimental a través de inhibición de expresión de mediadores proinflamatorios como RANTES, MCP-1 e IL-6.
Materiales y métodosProtocolo experimentalSe utilizaron ratas espontáneamente hipertensas (SHR) y Wistar-Kyoto (WKY), machos, de 14 semanas, proporcionados por el Instituto Venezolano de Investigaciones Científicas (IVIC) (Estado Miranda-Venezuela). Los animales se mantuvieron en un ambiente de temperatura controlada (22 a 24°C) con libre acceso a comida y agua. Se dividieron en 3 grupos experimentales (n=6 para cada grupo). El primer grupo (SHR-pCMV-HGF), fue tratado con el gen del HGF humano previamente clonado en el sitio BamHI del plásmido pcDNA3.1 (1mg/kg; Invitrogen). El segundo grupo (SHR-pcDNA3.1), recibió el vector vacío, y el tercer grupo (WKY) fue el grupo control. El tratamiento se administró a través de inyecciones hidrodinámicas en la vena dorsal de la cola durante 6 semanas. La presión arterial de los animales se midió semanalmente en los 3 grupos. El sacrificio se realizó 5 días después de la última aplicación del gen o del vector vacío, posteriormente se practicó laparotomía media y se extrajo la aorta; las secciones de este tejido se conservaron en solución salina fisiológica a 4°C.
Medición de la presión arterialLa medición de la presión arterial se realizó en ratas conscientes, con métodos electrónicos computarizados para la toma de la presión arterial en ratas (Life Science Instruments. Modelo 229 IITC INC). Los animales fueron aclimatados antes de comenzar cada medición a una temperatura de 28 a 29°C y se hizo una medición por semana. Este método permite medir la presión y el pulso a través de sensores que detectan pulsaciones en la cola del animal. Se realizaron 3 lecturas de la presión sanguínea de cada animal y se obtuvo una media por animal; al final se obtuvo un promedio por cada grupo experimental.
Preparación del plásmidoLa preparación del plásmido se realizó como se ha descrito previamente21. Brevemente, el plásmido de expresión pcDNA3.1 (Invitrogen) que contiene el gen HGF humano clonado en los sitios BamHI fue generosamente donado por el Dr. Youhua Liu (University of Pittsburgh, School of Medicine, Pennsylvania, EE. UU.). El plásmido fue transformado en bacterias JM-109 y seleccionadas en placas de agar con ampicilina. Los clones positivos fueron amplificados en medio Luria-Bertani. El plásmido fue purificado usando el kit PureYield Plasmid Maxiprep System (Promega,EE. UU.) siguiendo el protocolo del fabricante. La pureza y concentración del plásmido aislado fue confirmada mediante electroforesis en geles de agarosa y por espectrofotometría.
Reacción en cadena de la polimerasa en tiempo realSe extrajo el ARN total a partir de la aorta utilizando un kit comercial (SV Total ARN isolation system, Promega) siguiendo las instrucciones del fabricante y para la producción de ADN complementario se utilizó el kit comercial (Access RT-PCR system, Promega). Los niveles de expresión de MCP-1, RANTES, IL-6 fueron determinados usando los siguientes cebadores específicos sintetizados por Eurogentec: MCP-1 (sentido) 5‘-AGC CCA GAA ACC AGC CAA CTC-3’ y (anti-sentido) 5‘-GCC GAC TCA TTG GGA TCA TCT T-3‘, RANTES (sentido) 5‘-ATG AAG ATC TCT GCA GCT GCA TCC-3’ y (anti-sentido) 5‘-CTA GCT CAT CTC CAA ATA GTT G-3‘, IL-6 (sentido) 5‘-GAC TGA TGT TGT TGA CAG CCA GTG C-3’ (anti-sentido) 5‘-TAG CCA CTC CTT CTG TGA CTC TAA CT-3‘, HGF (sentido) 5‘-CCA GCT AGA AAC AAA GAC TTG AAA GA-3’ y (anti-sentido) 5‘-GAA ATG TTT AAG ATC TGT TTG CGT T-3’ y β-actin (sentido) 5‘-CAG CTG AGA GGG AAA TCG TG-3’ y (anti-sentido) 5‘-CGT TGC CAA TAG TGA TGA CC-3‘, utilizando SYBR green (Bio-Rad Laboratories, Richmond Cal, EE. UU.) en un termociclador IQ5 Multicolor Real time PCR Detection System (Bio-Rad Laboratories, Richmond Cal, EE. UU.). El gen de referencia a utilizar fue el de la subunidad 18S del ribosoma.
Preparación de la fracción nuclearUna porción de tejido de la aorta fue cortada en trozos muy pequeños, suspendida en solución tampón A (25mM Tris-HCl, pH 7,4, 130mM NaCl and 5mM KCl), para luego ser homogenizada. A continuación se centrifugó el homogeneizado a 7.000×g por 2min a 4°C. El sedimento obtenido fue resuspendido y lisado en solución tamponadora B fría (10mM Hepes, pH 7,9, 10mM KCl, 0,1mM EDTA, 0,1mM EGTA, 1mM PMSF y 1mM dithiothreitol [DTT]) durante 20min en frío. Luego se le agregó 100μl de Nonidet P-40 y se agitó vigorosamente en el vórtex para ser luego centrifugado a 12.000g durante 7min a 4°C. Seguidamente se extrajeron los núcleos con 100μl de tampón C frío (20mM HEPES, pH7,9, 400mM NaCl, 1mM EDTA, 1mM EGTA, 1mM PMSF y 1mM DTT), se incubó sobre hielo durante 2h y se centrifugó a 12.000×g por 7min a 4°C.
Western blotLas corridas se realizaron en minicámaras para electroforesis (Miniprotean III, Bio-Rad Laboratories, Richmond Cal, EE. UU.) utilizando geles de poliacrilamida al 12% y corridos a 80-120mV. Se empleó como tampón de corrida Tris 25mM, glicina 192mM, SDS 0,1%. Previo a esto las muestran fueron disueltas en la mezcla de disociación, a una concentración final de Tris-HCl 0,012M, pH6,8, SDS 0,4%, glicerol 5%, 2-mercaptoetanol 2.88mM, azul de bromofenol 0,02%. La mezcla fue calentada a 100°C por 5min, y se usaron marcadores de peso molecular de amplio rango de Bio-Rad Laboratories. Después de la corrida electroforética el gel de poliacrilamida se equilibraba en el gel en tampón de transferencia (glicina 192mM, metanol 20%, Tris 25mM, pH8,3) a 4°C durante 30min. Posteriormente, el gel y la membrana de nitrocelulosa fueron ensambladas en la celda de electrotransferencia, compatible con la minicámara de electroforesis. Antes de proceder a la reacción con los anticuerpos, la membrana de nitrocelulosa fue bloqueaba con una solución de albúmina sérica bovina al 5%, tween 20 al 0,5% en PBS por 1h a 24°C. Luego, la nitrocelulosa fue incubada con la dilución apropiada del anticuerpo primario (anti-MCP-1, anti-RANTES, anti-IL-6 o anti NF-kB) en solución de bloqueo, por 24horas a 4°C, seguidamente las membranas fueron lavadas con tween 20 al 0,1% en PBS, 3 veces durante 20min y después se incubo por 2h a temperatura ambiente con el respectivo anticuerpo secundario ligado a peroxidasa diluido en solución de bloqueo. Finalmente, las membranas fueron lavadas 3 veces con tween 20 al 0,1% en PBS y se reveladas usando como sustrato DAB (3,3-diaminobenzidina 1mM, H2O2 0,003% en PBS).
Metodología estadísticaLos resultados se expresan como la media±desviación estándar de los grupos estudiados. Los resultados fueron comparados por la prueba de ANOVA con postest de Tukey. Se solicitó y se obtuvo aprobación institucional para la realización de este estudio en animales de experimentación.
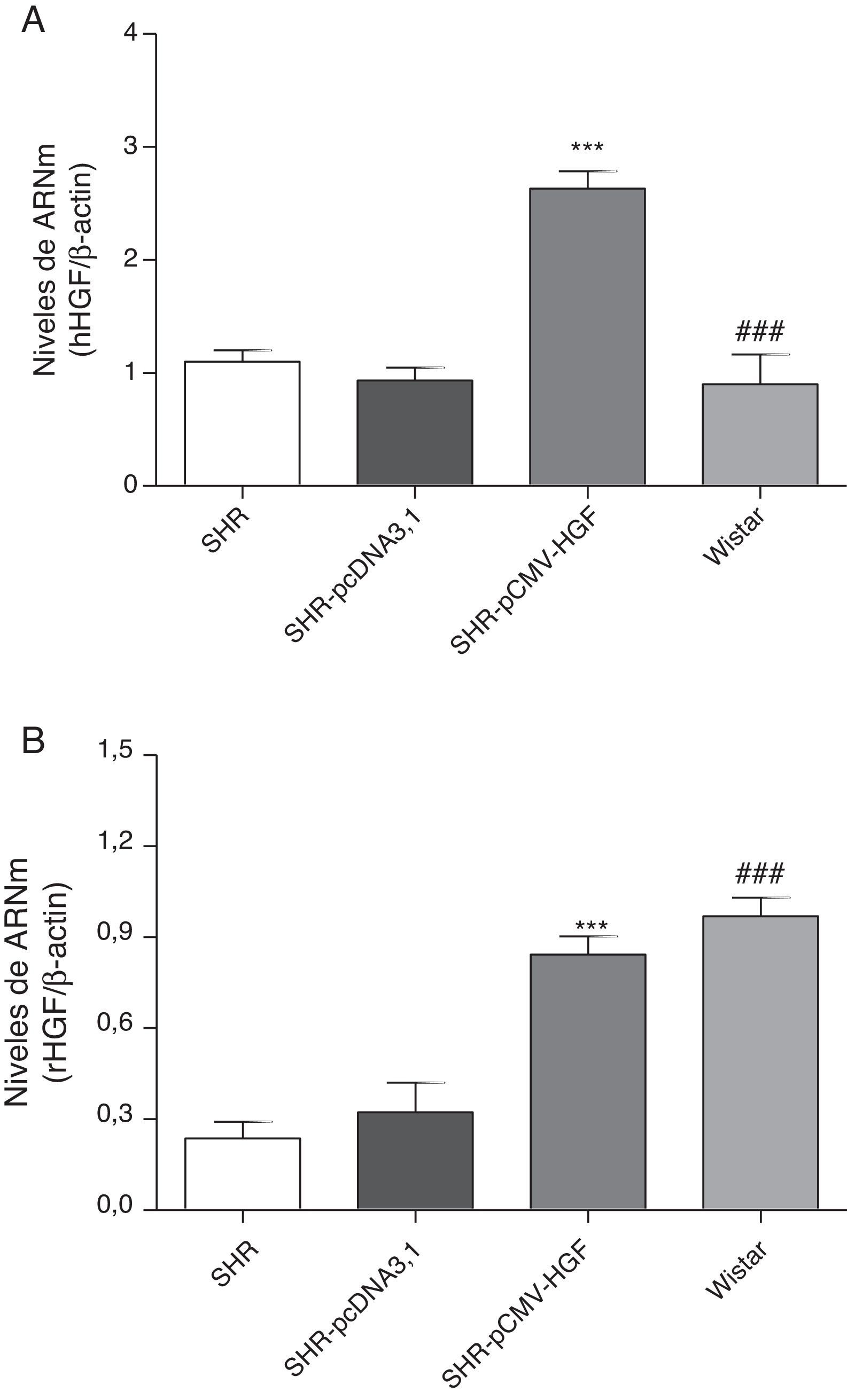
ResultadosExpresión del gen del factor de crecimiento del hepatocito humano y de ratas en la aorta de ratas espontáneamente hipertensasComo se observa en la figura 1A, la terapia génica fue capaz de inducir significativamente los niveles de expresión del gen humano en el grupo SHR-pCMV-HGF, en comparación con las ratas hipertensas no tratadas y con las ratas Wistar. Además, dicha terapia restituyó los niveles de expresión del gen a valores similares a los observados en las ratas controles (fig. 1B).
, del grupo SHR-pcDNA3.1 (n=6) y grupo control que fueron sacrificadas al final de la sexta semana; se determinó la expresión del gen del HGF de humano (hHGF) por RT-qPCR. Los valores representan la media±la desviación estándar de las ratas evaluadas por grupo. Se evaluó cada muestra por triplicado, por rata. B. Biopsias de ratas del grupo SHR-pCMV-HGF (n=6), del grupo SHR-pcDNA3.1 (n=6) y grupo control que fueron sacrificadas al final de la sexta semana; se determinó la expresión del gen del HGF de rata (rHGF) por RT-qPCR. Los valores representan la media±la desviación estándar de las ratas evaluadas por grupo. Se evaluó cada muestra por triplicado, por rata. **p<0,01 comparación entre los grupos SHR-pCMV-HGF y SHR-pcDNA3.1. ###p<0,01 comparación entre los grupos SHR-pCMV-HGF y Wistar.")
Expresión del HGF en SHR tratadas y no tratadas con hHGF y en ratas WKY.
A. Biopsias de ratas del grupo SHR-pCMV-HGF (n=6), del grupo SHR-pcDNA3.1 (n=6) y grupo control que fueron sacrificadas al final de la sexta semana; se determinó la expresión del gen del HGF de humano (hHGF) por RT-qPCR. Los valores representan la media±la desviación estándar de las ratas evaluadas por grupo. Se evaluó cada muestra por triplicado, por rata.
B. Biopsias de ratas del grupo SHR-pCMV-HGF (n=6), del grupo SHR-pcDNA3.1 (n=6) y grupo control que fueron sacrificadas al final de la sexta semana; se determinó la expresión del gen del HGF de rata (rHGF) por RT-qPCR. Los valores representan la media±la desviación estándar de las ratas evaluadas por grupo. Se evaluó cada muestra por triplicado, por rata.
**p<0,01 comparación entre los grupos SHR-pCMV-HGF y SHR-pcDNA3.1.
###p<0,01 comparación entre los grupos SHR-pCMV-HGF y Wistar.
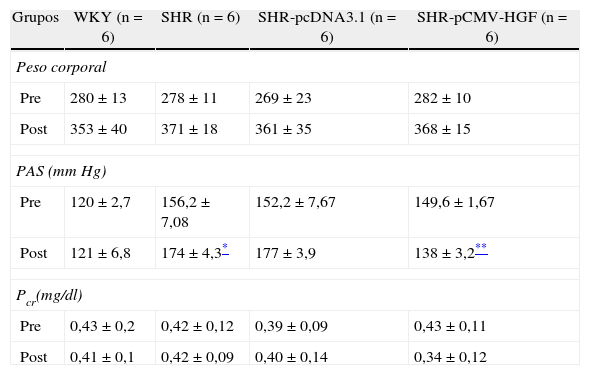
Como se observa en la tabla 1 las ratas WKY mantuvieron valores de presión arterial normal durante todo el experimento. Por otra parte, en las ratas SHR se observaron valores de presión arterial significativamente mayores al inicio del experimento. Tanto en el grupo de ratas SHR no inyectadas, como en el grupo inyectado con el vector vacío, se observó un aumento adicional de la presión arterial durante todo el experimento. En contraste, en el grupo de ratas SHR que recibieron la terapia génica con el HGF disminuyó la presión sistólica en SHR-pCMV-HGF vs SHR-pcADN3.1 (140±3,3 vs 170,3mmHg, p<0,01).
Valores de peso corporal, presión arterial y creatinina plasmática en SHR tratadas y no tratadas con HGF y en ratas WKY
| Grupos | WKY (n=6) | SHR (n=6) | SHR-pcDNA3.1 (n=6) | SHR-pCMV-HGF (n=6) |
| Peso corporal | ||||
| Pre | 280±13 | 278±11 | 269±23 | 282±10 |
| Post | 353±40 | 371±18 | 361±35 | 368±15 |
| PAS (mmHg) | ||||
| Pre | 120±2,7 | 156,2±7,08 | 152,2±7,67 | 149,6±1,67 |
| Post | 121±6,8 | 174±4,3* | 177±3,9 | 138±3,2** |
| Pcr(mg/dl) | ||||
| Pre | 0,43±0,2 | 0,42±0,12 | 0,39±0,09 | 0,43±0,11 |
| Post | 0,41±0,1 | 0,42±0,09 | 0,40±0,14 | 0,34±0,12 |
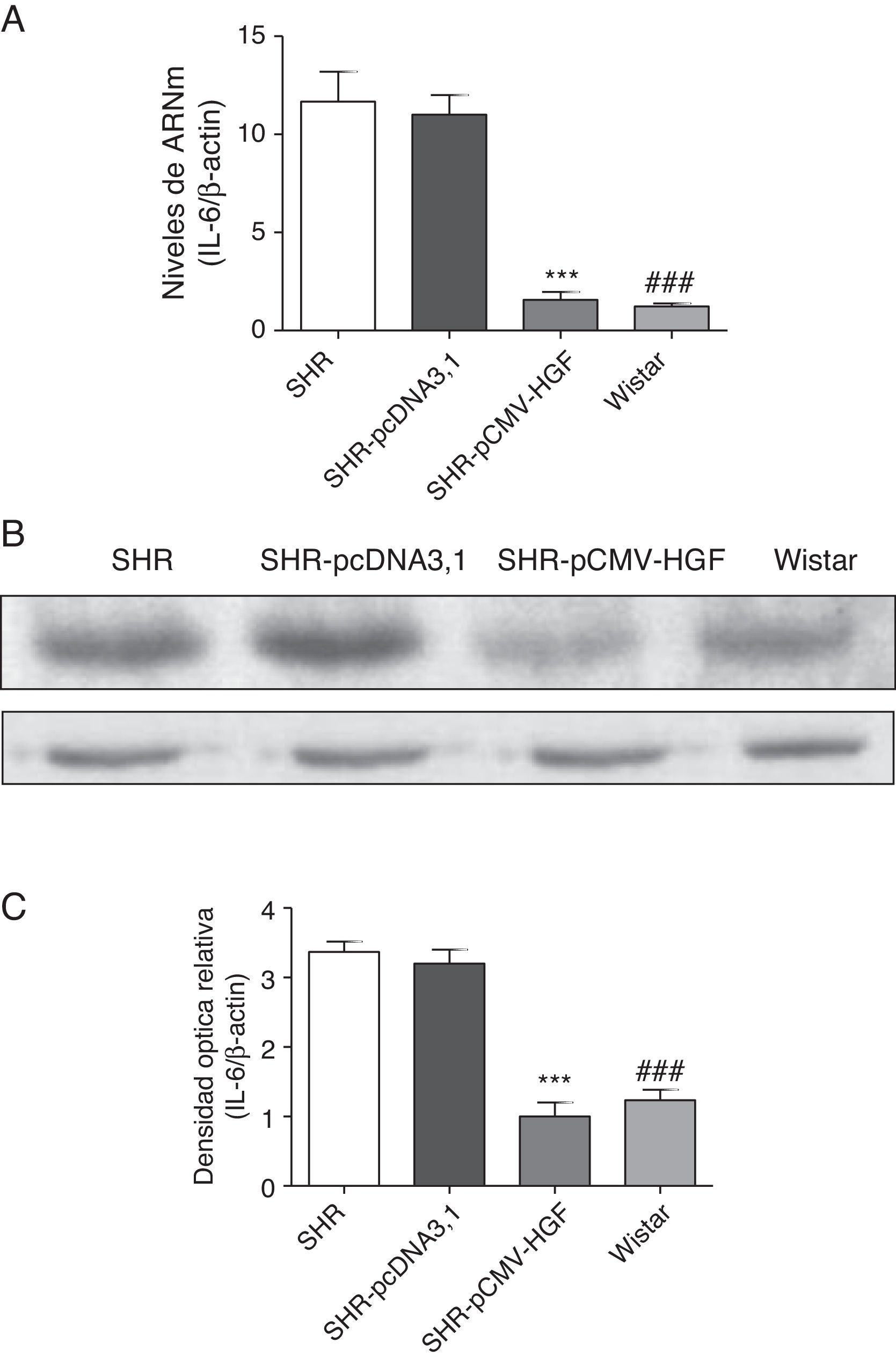
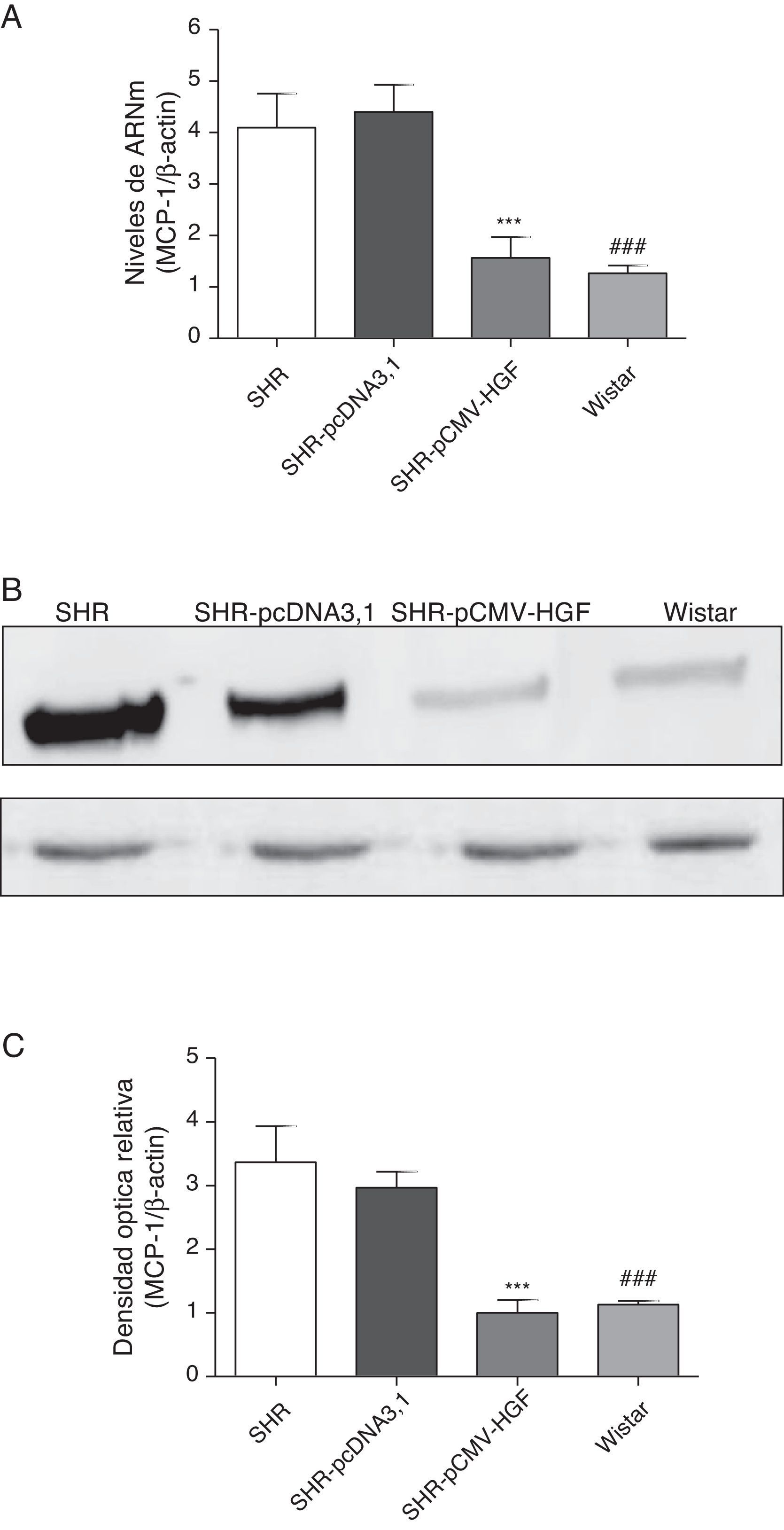
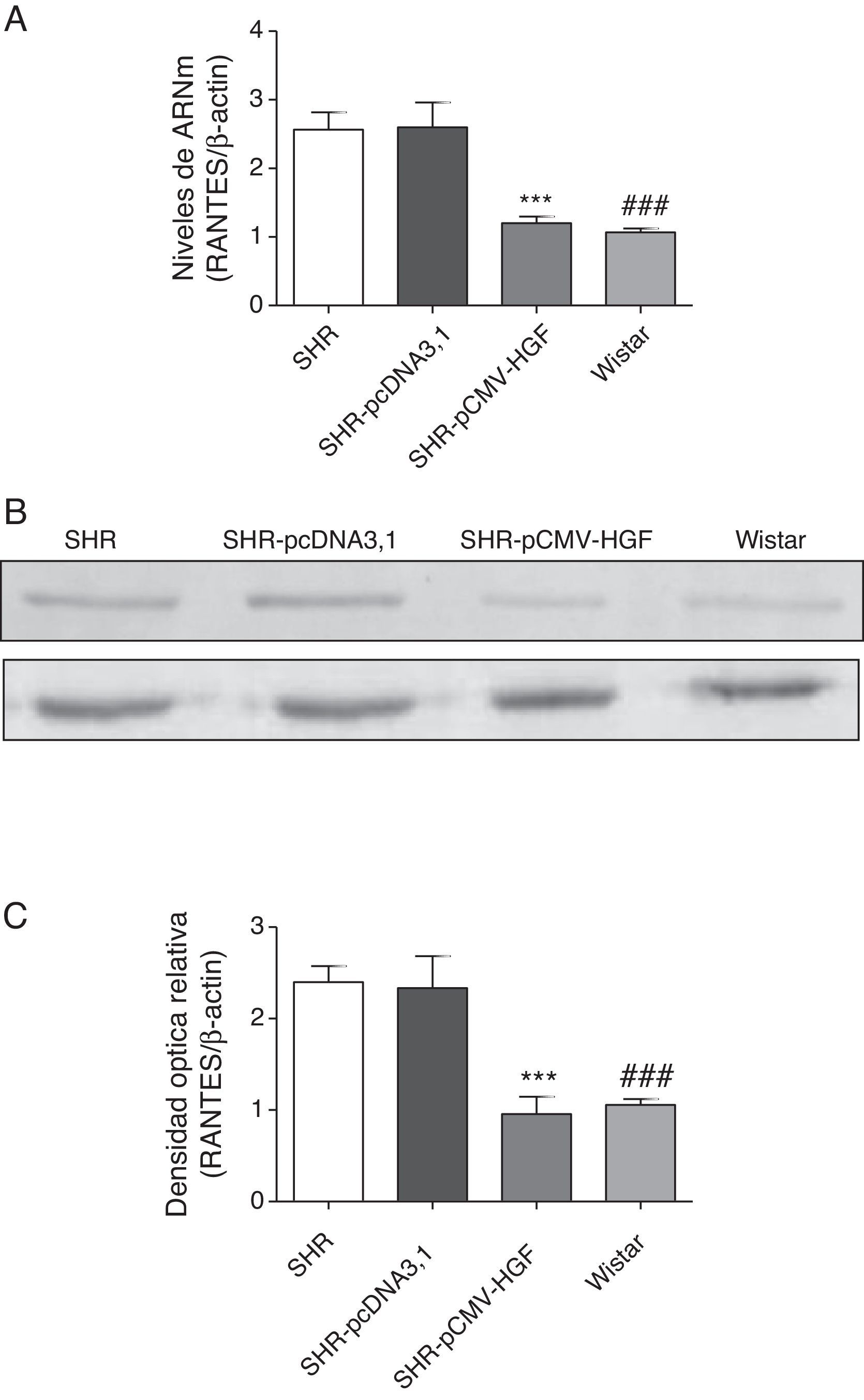
Previamente se ha demostrado que el HGF es capaz de ejercer un efecto antiinflamatorio al interferir los mecanismos de señalización mediados por el factor de transcripción NF-kB posterior al daño22. En este estudio se analizó el efecto de la terapia génica con el HGF en la expresión de las citocinas proinflamatorias, IL-6, MCP-1 y RANTES (figs. 2–4 respectivamente) en aorta de ratas SHR y WKY. El análisis por RT-qPCR y Western blot demostró una disminución significativa de la expresión de IL-6, a nivel de ARNm y proteico en la aorta de las ratas SHR inyectadas con el plásmido pCMV-HGF, al compararlas con el grupo SHR-pcDNA3.1 (figs. 2A y B). Del mismo modo, la expresión proteica y del ARNm de MCP-1 estuvo disminuida en la aorta en el grupo tratado con el gen del HGF en relación con el grupo tratado con el vector vacío (p<0,01) (figs. 3A y B). La expresión de RANTES disminuyó significativamente en la aorta en el grupo SHR-pCMV-HGF, al compararlas con el grupo SHR-pcDNA3.1. (p<0.01) (figs. 4A y B). En conjunto, la expresión de las citocinas proinflamatorias no fue diferente entre los grupos SHR-pCMV-HGF y el grupo control.
, del grupo SHR-pcDNA3.1 (n=6) y grupo control que fueron sacrificadas al final de la sexta semana. A. Se determinó la expresión de ARNm de IL-6 en aorta por RT-qPCR. Los valores representan la media±la desviación estándar de las ratas evaluadas por grupo. Se evaluó cada muestra por triplicado, por rata. B. Representa la expresión de IL-6 determinada por Western blot en SHR, SHR-pcDNA3.1, SHR-HGF and Wistar, n=6 en cada grupo. C. Representa la densidad óptica relativa de la expresión de IL-6 en SHR, SHR-pcDNA3.1, SHR-HGF y Wistar, n=6 en cada grupo. ***p<0,01 comparación entre los grupos SHR-pCMV-HGF y SHR-pcDNA3.1. ###p<0,01 comparación entre los grupos SHR y Wistar.")
Expresión de IL-6 en aorta de ratas SHR tratadas y no tratadas con HGF.
Biopsias de ratas del grupo SHR-pCMV-HGF (n=6), del grupo SHR-pcDNA3.1 (n=6) y grupo control que fueron sacrificadas al final de la sexta semana. A. Se determinó la expresión de ARNm de IL-6 en aorta por RT-qPCR. Los valores representan la media±la desviación estándar de las ratas evaluadas por grupo. Se evaluó cada muestra por triplicado, por rata.
B. Representa la expresión de IL-6 determinada por Western blot en SHR, SHR-pcDNA3.1, SHR-HGF and Wistar, n=6 en cada grupo.
C. Representa la densidad óptica relativa de la expresión de IL-6 en SHR, SHR-pcDNA3.1, SHR-HGF y Wistar, n=6 en cada grupo.
***p<0,01 comparación entre los grupos SHR-pCMV-HGF y SHR-pcDNA3.1.
###p<0,01 comparación entre los grupos SHR y Wistar.
, del grupo SHR-pcDNA3.1 (n=6) y grupo control que fueron sacrificadas al final de la sexta semana. A. Se determinó la expresión de ARNm de MCP-1 en aorta por RT-qPCR. Los valores representan la media±la desviación estándar de las ratas evaluadas por grupo. Se evaluó cada muestra por triplicado, por rata. B. Representa la expresión de MCP-1 determinada por Western blot en SHR, SHR-pcDNA3.1, SHR-HGF and Wistar, n=6 en cada grupo. C. Representa la densidad óptica relativa de la expresión de MCP-1 en SHR, SHR-pcDNA3.1, SHR-HGF y Wistar, n=6 en cada grupo. ***p<0,01 comparación entre los grupos SHR-pCMV-HGF y SHR-pcDNA3.1. ###p<0,01 comparación entre los grupos SHR y Wistar.")
Expresión de MCP-1 en aorta de ratas SHR tratadas y no tratadas con HGF.
Biopsias de ratas del grupo SHR-pCMV-HGF (n=6), del grupo SHR-pcDNA3.1 (n=6) y grupo control que fueron sacrificadas al final de la sexta semana.
A. Se determinó la expresión de ARNm de MCP-1 en aorta por RT-qPCR. Los valores representan la media±la desviación estándar de las ratas evaluadas por grupo. Se evaluó cada muestra por triplicado, por rata.
B. Representa la expresión de MCP-1 determinada por Western blot en SHR, SHR-pcDNA3.1, SHR-HGF and Wistar, n=6 en cada grupo.
C. Representa la densidad óptica relativa de la expresión de MCP-1 en SHR, SHR-pcDNA3.1, SHR-HGF y Wistar, n=6 en cada grupo.
***p<0,01 comparación entre los grupos SHR-pCMV-HGF y SHR-pcDNA3.1.
###p<0,01 comparación entre los grupos SHR y Wistar.
, del grupo SHR-pcDNA3.1 (n=6) y grupo control que fueron sacrificadas al final de la sexta semana. Se determinó la expresión de ARNm de RANTES en aorta por RT-qPCR. Los valores representan el promedio+la desviación estándar de las ratas evaluadas por grupo. Se evaluó cada muestra por triplicado, por rata. B. Representa la expresión de RANTES determinada por Western blot en SHR, SHR-pcDNA3.1, SHR-HGF and Wistar, n=6 en cada grupo. C. Representa la densidad óptica relativa de la expresión de RANTES en SHR, SHR-pcDNA3.1, SHR-HGF y Wistar, n=6 en cada grupo. ***p<0,01 comparación entre los grupos SHR-pCMV-HGF y SHR-pcDNA3.1. ###p<0,01 comparación entre los grupos SHR y Wistar.")
Expresión de RANTES en aorta de ratas SHR tratadas y no tratadas con HGF.
A. Biopsias de ratas del grupo SHR-pCMV-HGF (n=6), del grupo SHR-pcDNA3.1 (n=6) y grupo control que fueron sacrificadas al final de la sexta semana. Se determinó la expresión de ARNm de RANTES en aorta por RT-qPCR. Los valores representan el promedio+la desviación estándar de las ratas evaluadas por grupo. Se evaluó cada muestra por triplicado, por rata.
B. Representa la expresión de RANTES determinada por Western blot en SHR, SHR-pcDNA3.1, SHR-HGF and Wistar, n=6 en cada grupo.
C. Representa la densidad óptica relativa de la expresión de RANTES en SHR, SHR-pcDNA3.1, SHR-HGF y Wistar, n=6 en cada grupo.
***p<0,01 comparación entre los grupos SHR-pCMV-HGF y SHR-pcDNA3.1.
###p<0,01 comparación entre los grupos SHR y Wistar.
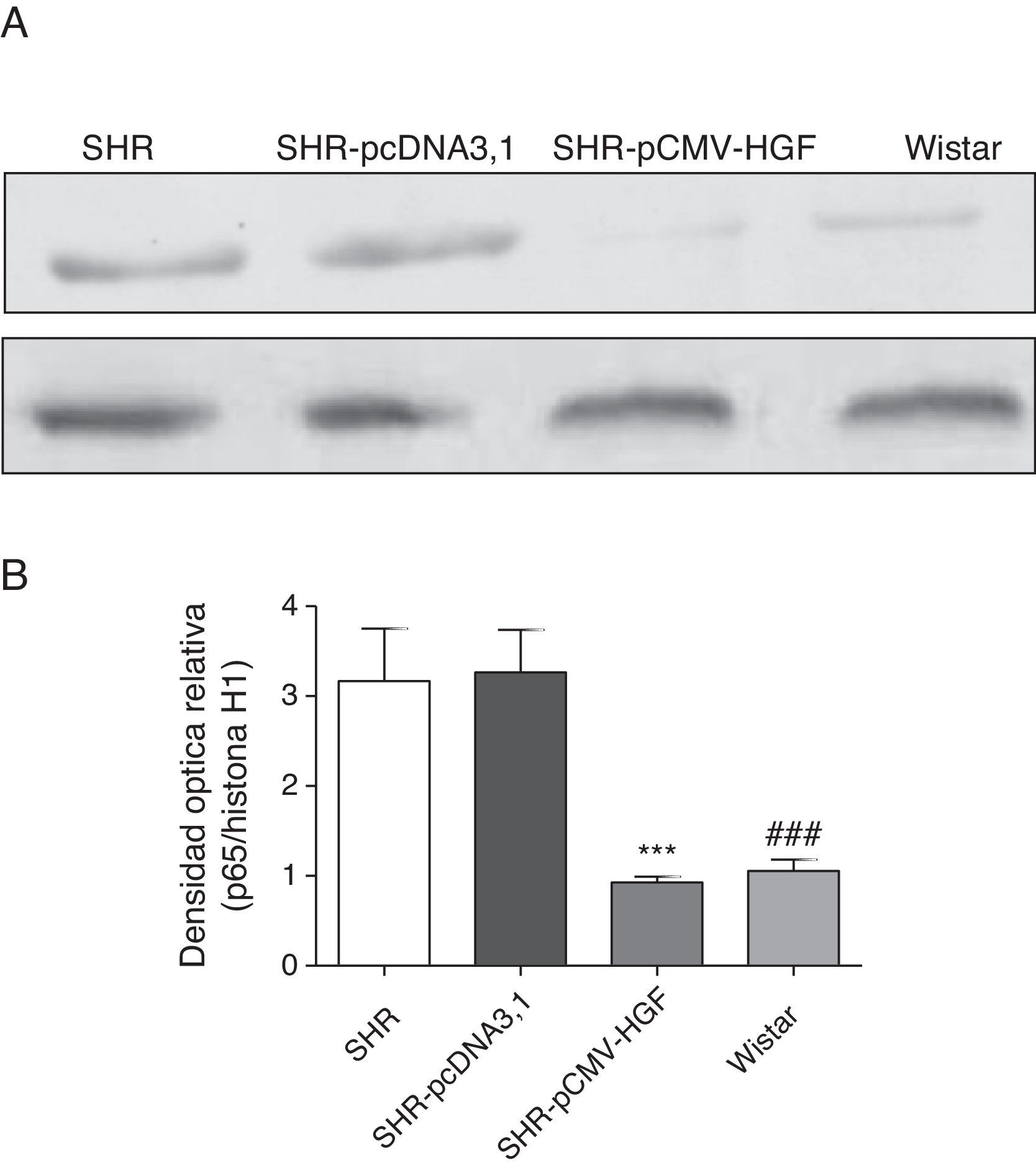
Como se observa en la figura 5, la subunidad p65 del factor de transcripción NF-κB se encontró aumentada en la fracción nuclear de las ratas hipertensas no tratadas y en el grupo inyectado con el vector vacío. Por otra parte, la terapia génica con HGF impidió el aumento de la localización nuclear del mencionado factor de transcripción.
, del grupo SHR-pcDNA3.1 (n=6) y grupo control que fueron sacrificadas al final de la sexta semana. Se determinó la expresión de la subunidad p65 del factor de transcripción NF-κB. Los valores representan la media+la desviación estándar de las ratas evaluadas por grupo. Se evaluó cada muestra por triplicado, por rata. B. Representa la densidad óptica relativa de la subunidad p65 del factor de transcripción NF-κB en los grupos estudiados. ***p<0,01 comparación entre los grupos SHR-pCMV-HGF y SHR-pcDNA3.1. ###p<0,01 comparación entre los grupos SHR y Wistar.")
Expresión de la subunidad p65 del factor de transcripción NF-κB en aorta de ratas SHR tratadas y no tratadas con HGF.
A. Biopsias de ratas del grupo SHR-pCMV-HGF (n=6), del grupo SHR-pcDNA3.1 (n=6) y grupo control que fueron sacrificadas al final de la sexta semana. Se determinó la expresión de la subunidad p65 del factor de transcripción NF-κB. Los valores representan la media+la desviación estándar de las ratas evaluadas por grupo. Se evaluó cada muestra por triplicado, por rata.
B. Representa la densidad óptica relativa de la subunidad p65 del factor de transcripción NF-κB en los grupos estudiados.
***p<0,01 comparación entre los grupos SHR-pCMV-HGF y SHR-pcDNA3.1.
###p<0,01 comparación entre los grupos SHR y Wistar.
Existe una creciente evidencia que indica que los mecanismos inflamatorios tienen un rol fundamental en la fisiopatología de la HTA2. Por otra parte, estudios recientes sugieren que el HGF suprime la inflamación aguda y crónica en una variedad de órganos y sistemas en modelos animales12. En el presente estudio se utilizó un esquema de terapia génica con el gen del factor de crecimiento de los hepatocitos en SHR. Se demostraron niveles de expresión significativos de dicho gen en la aorta de las ratas tratadas, asociado con una disminución de la expresión de mediadores proinflamatorios y de la activación del factor nuclear NF-kB, así como una reducción de la HTA en comparación con el grupo control. Estudios previos han demostrado que las concentraciones séricas del HGF en SHR son significativamente más altas que en las ratas normotensas Wistar Kyoto. Por el contrario, en tejidos como el corazón, la aorta y el riñón la expresión del HGF se encontró disminuida en comparación con la cepa WKY23. Por otra parte, varios estudios han relacionado la HTA con inflamación en modelos experimentales como las ratas Dahl, SHR y el modelo de infusión de Ang II24–26. Estudios previos sugieren un papel importante del estrés oxidativo vascular en SHR27, lo cual podría contribuir a aumentar la señalización intracelular como consecuencia de la activación del NF-kB28. De hecho, el bloqueo de la señalización del NF-kB y la producción de ROS tiene un efecto antiinflamatorio acompañado de disminución de la presión arterial29.
En esta investigación se demostró que el HGF disminuye la expresión del ARNm IL-6, MCP-1, RANTES y la activación del NF-kB. La disminución del ARNm de IL-6 demostrada en estos animales podría sugerir, al menos en parte, que la acción del HGF sobre esta citocina se realiza ejerciendo un doble efecto: 1) al disminuir la expresión de IL-6 se reduce la activación del NF-kB, como se demostró en estudios previos, donde este factor activa genes mediadores de la inflamación y ha sido asociado al proceso inflamatorio vascular, renal y cardiaco30. Similarmente, el NF-kB ha sido involucrado en la acción proinflamatoria de la Ang II en otros modelos de hipertensión en ratas31, demostrándose además que su inhibición disminuye la inflamación vascular inducida por Ang II en ratas32; y 2) se ha relacionado la disminución de la IL-6 con reducción en la expresión del gen del receptor AT1 en tejido vascular, con un menor desarrollo y progresión del daño endotelial, similar a lo reportado con la administración de inhibidores de la enzima convertidora de angiotensina33. Los resultados de este estudio mostraron disminución de la IL-6 en el grupo SHR-pCMV-HGF, asociada a menor inflamación en la pared de la aorta, lo que probablemente podría estar relacionado con la menor actividad del NF-kB, quizás por un mecanismo involucrado con disminución del receptor AT1. En observaciones previas se asoció la HTA de SHR a procesos inflamatorios de la pared vascular34. Varios marcadores de la inflamación como ICAM-1, VCAM, MCP-1 e IL-6 se encontraron aumentados en la pared vascular de ratas hipertensas28, así como el incremento en la circulación de marcadores proinflamatorios en SHR35 y en pacientes hipertensos36. En conjunto, estos estudios demuestran el papel del proceso inflamatorio en la progresión del daño vascular asociado a hipertensión5, sugiriendo a su vez que la HTA está asociada con procesos inflamatorios generalizados mediados por la producción de factores proinflamatorios generados tanto en la pared vascular como a nivel extravascular9. En la actualidad el concepto de inflamación sistémica desempeña un papel central en el desarrollo de enfermedades vasculares, pues ha evolucionado en muy variadas direcciones, para explicar múltiples circunstancias en las que se ha comprobado la alteración histológica y funcional5. La relación entre la hipertensión y la inflamación sistémica representa una nueva área de investigación3. Evidencias recientes demuestran una compleja conexión entre la inflamación sistémica, la activación de las células vasculares y los cambios estructurales en las arterias. La inflamación y la hipertensión pueden interactuar la una con la otra de una manera bidireccional, determinando el desarrollo de ateroesclerosis y futuras complicaciones cardiovasculares4. Sin embargo, algunos estudios epidemiológicos demuestran que la presencia de un estatus de baja inflamación crónica puede anticipar el desarrollo futuro de hipertensión1. Esta observación sugiere que el incremento en los niveles plasmáticos de mediadores inflamatorios observados en pacientes hipertensos no puede ser atribuido solamente al daño vascular inducido por la alta presión arterial. Nuevas líneas de investigación estudian la posibilidad de un efecto patogénico directo de los mediadores inflamatorios en alterar los mecanismos regulatorios del tono vascular que induce el desarrollo de HTA. Desde este punto de vista, la HTA puede considerarse como la resultante de un proceso inflamatorio con remodelación y engrosamiento de las paredes vasculares, al que se asocia una repuesta inmunológica. De esta forma, se describe a lo largo de los vasos sanguíneos arteriales y venosos la concurrencia de células inflamatorias, que involucran la inmunidad innata y la inmunidad adaptativa2.
El HGF reduce la inflamación del intestino, atribuible a una acción antiinflamatoria directa9. Extensas investigaciones sobre el proceso inflamatorio destacan la complejidad de las interacciones entre mediadores solubles y moléculas de membranas en la célula endotelial, donde las citocinas con funciones quimotácticas desempeñan una función clave en la inmunomodulación, generando señales direccionales que ejercen un efecto directo en la migración de los leucocitos29. La quimiocina MCP-1 se encuentra aumentada en la pared vascular de conejos en un modelo de ateroesclerosis, donde es capaz de inducir la expresión de moléculas de adhesión y la secreción de IL-6, esta última implicada en la patogénesis de la ateroesclerosis37. Previamente se demostró en el riñón de ratas con HTA que la lesión del tejido está relacionada con el rol quimiotáctico del MCP-1, apoyando la relación que indica que los mecanismos inflamatorios contribuyen al daño en diferentes formas de hipertensión38. Esta afirmación se basa en estudios en los cuales tratamientos que disminuyen las concentraciones séricas de ARNm de marcadores inflamatorios se acompañaron de disminución de la presión arterial33.
Evidencias previas sugieren que el HGF suprime la activación del NF-kB, y la señalización implicada en el control de la expresión de varias citocinas proinflamatorias8. Recientemente se ha sugerido que la enzima glucógeno sintetasa cinasa (GSK-3β) está relacionada de manera importante en la transducción de la señalización del NF-kB y es esencial en su activación a través de un mecanismo dependiente de la vía de la PI 3-cinasa. El HGF, al suprimir la activación del NF-kB, bloquea la secuencia de eventos proinflamatorios e induce la inhibición de la activación de la GSK-3β. Algunos autores sugieren que la GSK-3β controla la activación del NF-kB por expresión directa del gen y que la inhibición de la GSK-3β suprime la expresión de algunas moléculas proinflamatorias que son inducidas por el NF-kB, incluyendo RANTES, MCP-1, IL-8 y la IL-639. Este mecanismo podría estar relacionado con los resultados obtenidos en este estudio, en el cual se demostró disminución de la expresión de citocinas pro-inflamatorias como IL-6, MCP-1 y RANTES en la aorta de los animales a los cuales se les administró terapia génica con HGF; estos resultados evidencian que la disminución del proceso inflamatorio en la pared de la aorta podría contribuir a una menor resistencia vascular en la misma. Todos estos efectos asociados, la disminución de las citocinas proinflamatorias y de la activación del NF-kB inducen una reducción de la inflamación en la arteria, y probablemente en los vasos sanguíneos de forma generalizada, con la consecuente disminución del daño tisular y quizás una menor resistencia vascular. En conjunto, estos mecanismos podrían explicar las cifras de presión arterial más bajas observadas en ratas SHR a las que se les administró terapia génica con HGF, sugiriendo que el efecto antiinflamatorio de este factor puede ejercer un efecto antihipertensivo.
ConclusiónEn conclusión, nuestros resultados indican que la reducción vascular del HGF en SHR promueve la activación de mediadores proinflamatorios y el desarrollo de HTA. Adicionalmente, este estudio demuestra que la terapia génica con HGF atenúa el desarrollo de HTA a través de la disminución en la expresión de IL-6, MCP-1 y RANTES vía inhibición de la activación del factor de transcripción NF-kB.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes y que todos los pacientes incluidos en el estudio han recibido información suficiente y han dado su consentimiento informado por escrito para participar en dicho estudio.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
FinanciaciónInstituto Venezolano de Investigaciones Científicas (IVIC-Zulia, Venezuela).
Conflicto de interesesLos autores de este trabajo declaramos que no existe conflicto de intereses.
AgradecimientosAgradecemos el aporte de la Dirección de Investigación de la Facultad de Medicina de la Universidad del Zulia.