


Los linfocitos T y B migran continuamente desde la sangre hacia los órganos linfoides secundarios, en los cuales antígenos procedentes de tejidos periféricos son procesados y presentados por células presentadoras. Tras permanecer en estos órganos temporalmente, de no producirse el reconocimiento antigénico, los linfocitos abandonan los mismos y son devueltos a la sangre para reiniciar este patrón migratorio conocido como recirculación linfocitaria. El continuo movimiento de los linfocitos a través de estos órganos incrementa la probabilidad de que se produzca el encuentro linfocito-antígeno específico, y es por tanto imprescindible para el eficaz desarrollo de respuestas inmunitarias adaptativas. Los principales factores reguladores del tráfico linfocitario son proteínas de la familia de las quimiocinas y el lípido esfingosina- 1-fosfato. Mediante la activación de receptores de membrana, estos mediadores promueven la adhesión de los linfocitos circulantes en el interior de vasos sanguíneos específicos, el acceso a los órganos linfoides, la migración en su interior, y la posterior salida a los conductos linfáticos o la sangre. El objetivo de esta revisión es repasar el estado actual del conocimiento de los mecanismos de señalización intracelular que son activados por estos receptores y que sustentan la continua recirculación linfocitaria.
Lymphocytes migrate from the blood stream into secondary lymphoid organs (SLO), where antigens (Ag) collected in the periphery are displayed on the surface of Ag-presenting cells. Continuous lymphocyte recirculation throughout SLO greatly increases the chances that rare Agspecific T and B cells encounter their cognate Ag, making it a prerequisite for effective immune surveillance. Members of the chemokine family of proteins, as well as the lipid mediator sphingosine-1-phosphate (S1P), are the main factors orchestrating dynamic trafficking of lymphocytes. They exert this control by activating specific receptors, which promote lymphocyte adhesion inside specific microvessels of SLO, subsequent migration inside lymphoid tissue and egress into the blood. This article covers our current understanding of the complex intracellular signaling mechanisms that are activated downstream of these receptors and contribute to lymphocyte recirculation.
Adaptive immune responses are elicited by lymphocytes, which are cells capable of mounting reactions against almost an infinite number of Ag in a highly specific fashion. The versatility of Ag recognition emerges during lymphocyte ontogeny, where unique genetic mechanisms generate a highly diverse pool of T and B lymphocytes, each of which bear on their surface a unique receptor recognizing a specific Ag(1). The diversity of this immune repertoire enables adaptive responses to cope with a huge array of different external insults at the expense of drastically limiting the number of lymphocytes specifically recognizing a given Ag. Therefore, to maximize the likelihood of the otherwise improbable antigenic encounter, lymphocytes have evolved to continuously migrate throughout the body, transiently residing in the so- called secondary lymphoid organs (SLO) that are strategically located throughout the organism(1-3). Continuous lymphocyte recirculation is regulated by members of the chemokine family and the lipid mediator S1P, which modulate both lymphocyte adhesive interactions inside vascular beds and migration into and within lymphoid tissues(4,5). This review aims to give an abbreviated overview on the signal transduction machinery activated by both chemokine and S1P receptors promoting lymphocyte migration or adhesion, and ultimately controlling lymphocyte trafficking.
MOLECULAR CUES FOR LYMPHOCYTE TRAFFICKING: CHEMOKINES AND S1PNaïve lymphocytes recirculate through SLO, which form an intricate network of lymphoid aggregates including Peripheral Lymph nodes (PLN), Spleen (SPL), Peyer's Patches (PP) and Mucose-associated Lymphoid Tissue (MALT). Ag present in peripheral tissues, blood or mucosal barriers are channeled to SLO which constantly recruit blood-borne lymphocytes and therefore serve as regional control centers for lymphocyte-antigen encounter. The hallmark of SLO is their defined microanatomical organization in subset specific compartments, T cell areas and B cell follicles, which provide the adequate environment for lymphocyte activation and survival. Inside these areas, T and B cells persistently screen the surface of tissue-derived antigen presenting cells. In absence of antigen recognition, after an average dwelling time of 8h for T cells and 12-16h for B cells, lymphocytes leave SLO returning to the blood to eventually migrate into a different SLO. Lymphocyte recirculation therefore involves homing of circulating T and B cells from blood to SLO, migration to specific regions within these organs, vigorous random motility while residing inside these areas, and finally, egress to blood to eventually reach other SLO.
Chemokines are a large family of small (8–12kDa) polypeptides originally described for their role in immune cell recruitment during inflammatory processes(5,6). In conjunction with the lipid S1P and other yet unknown factors, members of this protein family direct lymphocytes in their recirculation process. A mechanistic explanation on how this is achieved is included in this section.
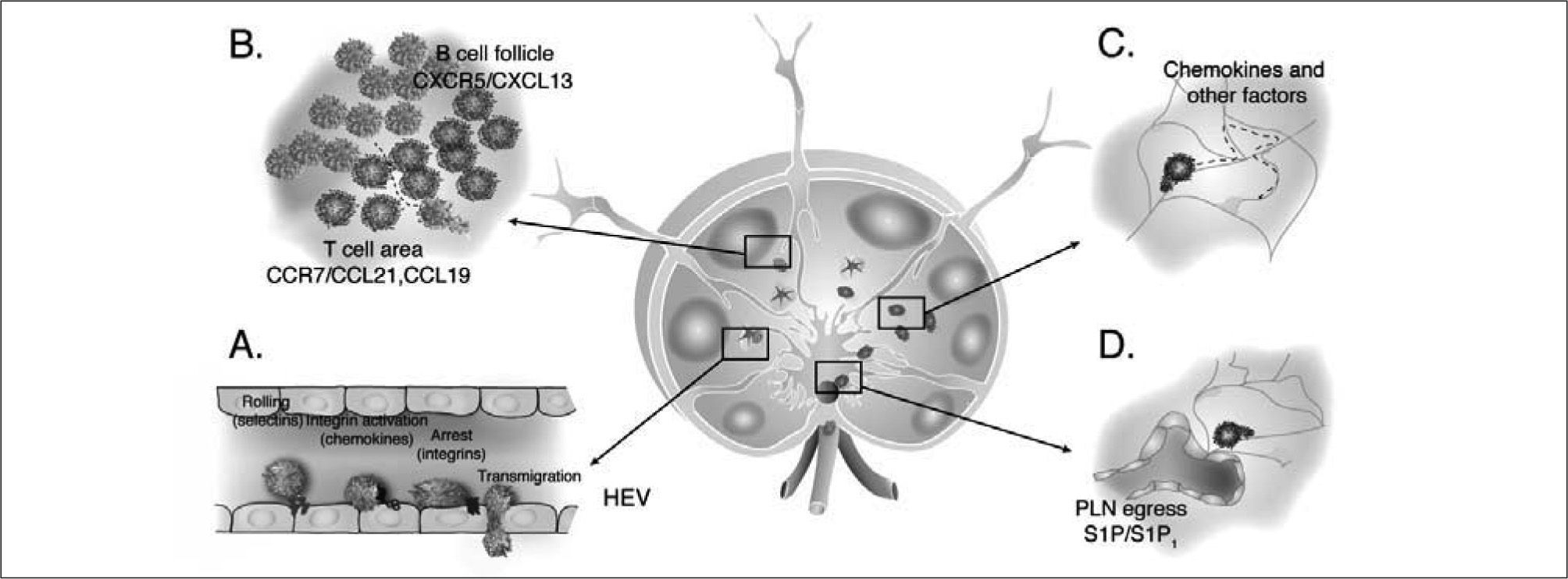
Lymphocyte entry to SLOLymphocytes enter PLN and PP via High Endothelial Venules (HEV), specialized microvessels expressing specific vascular determinants which favor selective recruitment of lymphocytes but not other blood cell types(1,7). Chemokines function as key regulators of the homing process, which occurs in discrete steps encompassing the sequential engagement of a series of receptor-ligand interactions(2). Inside HEV, blood-borne lymphocytes initiate a rolling movement along the vessel wall which slows them down and promotes a tighter contact with the endothelium, facilitating the interaction between HEV-presented chemokines and chemokine receptors (CKR). Binding of chemokines to CKR triggers rapid activation of adhesion molecules of the integrin family, promoting the arrest of rolling lymphocytes (Figure 1A)(8-10) which is followed by their passage across endothelium into interstitial space of SLO. Chemokine requirements for SLO entry slightly differ between T and B cells. T cell homing to PLN largely relies on CCR7 and its ligands CCL21/CCL19(11,12), as well as a minor contribution by CXCL12-CXCR4 interactions(13,14). Although B cell integrin activation is also primarily induced by CCR7 and CXCR4 in PLN, homing to PP additionally requires interaction of CXCL13 with the receptor CXCR5, whose expression is restricted to B cells and a subset of CD4+ T cells(14). Completion of the multi-step cascade culminates with transmigration of firmly adhered lymphocytes across the endothelium (Figure 1A). It was long thought that transmigration occurred in response to chemoattractant gradients(15). Nonetheless, this widely accepted notion was challenged by studies demonstrating that mere application of hydrodynamic shear flow forces on adhered lymphocytes suffices to promote diapedesis in vitro in presence of luminal but not basal chemokine(16). Hence, chemokines dictate lymphocyte homing to SLO by inducing lymphocyte firm adhesion to endothelium upon passage through specific microvascular beds.

Dynamics of lymphocyte migration inside PLN. A. Multi-step homing of lymphocytes takes place inside HEV. Lymphocytes in the bloodstream interact and roll along the endothelium of microvessels, a process mediated by selectin/selectin ligand interactions. Chemokines presented by endothelial cells, engage specific CKR in the surface of lymphocytes. Rapid intracellular signaling downstream of CKR triggers activation of integrins leading to arrest of rolling lymphocytes. Chemokine/CKR receptor requirements differ from T to B cells depending on the type of SLO but include CCL21/CCL19-CCR7, CXCL12-CXCR4 and CXCL13-CXCR5. B. T and B cell compartmentalization in T cell areas and B cell follicles relies on CCL21/CCL19 and CXCL13 expression, respectively. C. Lymphocytes randomly migrate inside these microenvironments in response to pro-migratory stimuli including chemokines and other yet unknown factors. D. PLN egress through sinuses located in the medulla of PLN requires SIP1 expression on lymphocytes, which probably migrate in response to S1P present in lymph.
Migration to splenic lymphoid tissue (white pulp) constitutes a remarkable exception since SPL lacks HEV. Nevertheless chemokines play an important role in guiding lymphocyte entry into white pulp cords(5). Circulating T and B cells are passively released into sinuses near the marginal zone, an area defining the border between the white pulp and the non-lymphoid tissue of the spleen (red pulp). While some cells are passively swept into venous sinusoids and continue their recirculation, others actively migrate into the white pulp in a chemokine-dependent fashion. T cell migration is directed by the expression of CCR7 ligands, while B cells are attracted and transiently retained in T cell specific areas by CCR7 ligands, to later migrate into B cell follicles in response to CXCL13(5,17,18).
Interstitial migrationOnce inside SLO, T and B cells readily segregate into specific microenvironments. This characteristic anatomical organization is dependent on the selective expression of CCL21/CCL19 in T cell areas and CXCL13 in B cell follicles (Figure 1B)(17-20). The initial hypothesis postulating that lymphocytes were directed to these areas by sensing longrange soluble chemokine gradients was challenged when direct visualization of lymphocyte behavior inside intact tissues was achieved through Multi-photon Microscopy (MPM). Inside PLN stroma, lymphocytes migrate vigorously with no apparent directionality, following the typical parameters of random walk (Figure 1C)(21-25). Such basal motility has been described to only partially rely on CCR7, requiring the contribution of other yet undefined GPCR-mediated signals(26,27).
Egress from lymphoid organsIn absence of inflammation, naive lymphocytes leave PLN via lymphatic vessels after an average dwell time of 8h in the case of T cells and 16h for B cells. Efferent lymphatics drain into the thoracic duct which releases lymph borne cells back into the blood circulation(3,4). Recent studies demonstrated that PLN egress signals guiding lymphocytes into lymphatic vessels are provided by S1P, a lipid mediator which is abundant in blood and lymph but present at low concentrations in lymphoid tissue (Figure 1D). Lack of expression of one of the five S1P receptors, S1P1, renders lymphocytes unresponsive to exit cues, thus clearing them from blood and lymph due to their sequestration in SLO(28). A model has been proposed by which the cyclical modulation of S1P1 expression by environmental S1P concentrations regulates lymphocyte transit through SLO. Lymphocytes express low S1P1 levels when isolated from blood and lymph where S1P levels are high. During lymphocyte dwelling in SLO, where concentrations of S1P are low, S1P1 expression gradually increases. According to this model, recovery of S1P responsiveness enables directed migration along an S1P gradient towards lymphatics antagonizing retention signals originated by the expression of CCR7 ligands in lymphoid tissue(29,30).
INTRACELLULAR SIGNALING PATHWAYS CONTROLING LYMPHOCYTE TRAFFICKINGLymphocyte migration within lymphoid tissue, is a highly complex process which requires the sensing of directionality towards a chemotactic stimulus, cell polarization, and cellular translocation. Asymmetrical shape changes are accompanied by the formation of two defined structures at the front and rear of the cell, namely the leading edge and trailing edge or uropod(31). The propelling force for cell movement is then provided through emission of protrusions at the leading edge followed by contraction of the uropod in a synchronized reiterative manner.
Although both migration and adhesion have been historically studied in model organisms, cell lines or neutrophils, it became recently clear that substantial mechanistic differences exist even between closely related cell types(32). How CKR and S1P1 integrate environmental signals to elicit these functional outcomes in T and B cells is therefore incompletely understood to date. The following section reviews the advancements in our understanding on the signaling pathways regulating lymphocyte trafficking with special emphasis on in vivo evidence derived from genetically-engineered mouse models. Of note, although in certain cell types adhesion and migration are interdependent processes, experimental evidence suggests that in lymphocytes they occur in a largely independent manner(33), implying the presence of separate signaling networks. Thus, both events are analyzed independently in this review. Finally, although the signaling events described in this section are interconnected, for reasons of clarity they will be divided in: i) membraneproximal events ii) cytoplasmic signaling events related to small GTPases and the cytoskeleton.
Membrane-proximal signaling eventsGPCR couple on their intracellular side to G proteins, heterotrimeric complexes formed by a Gα subunit and a Gβγ dimer (Figure 2). The Gα subunit has intrinsic GTPase activity and binds GDP in the resting state. Upon ligand binding to GPCR, Gα subunit changes GDP for GTP and rapidly dissociates from the Gβγ dimer, eliciting numerous signaling pathways(34). The CKR and S1P receptors involved in lymphocyte recirculation are mostly Gαi-coupled (CCR7, CXCR5, CXCR4 and S1P1). Selective blocking of Gαi-coupled receptor signaling after treatment with pertussis toxin almost completely abrogates lymphocyte adhesion inside HEV and migration into splenic white pulp in vivo(9,13,35,36). Likewise, lymphocyte intranodal migration is promoted through Gαi-induced pathways which are known to partially emanate from CCR7 activation, but mostly derive from other yet unknown chemokinetic signals. Genetic studies have demonstrated that Gαi2 is the main Gαi isoform regulating T and B cell signaling through CKR(37,38).
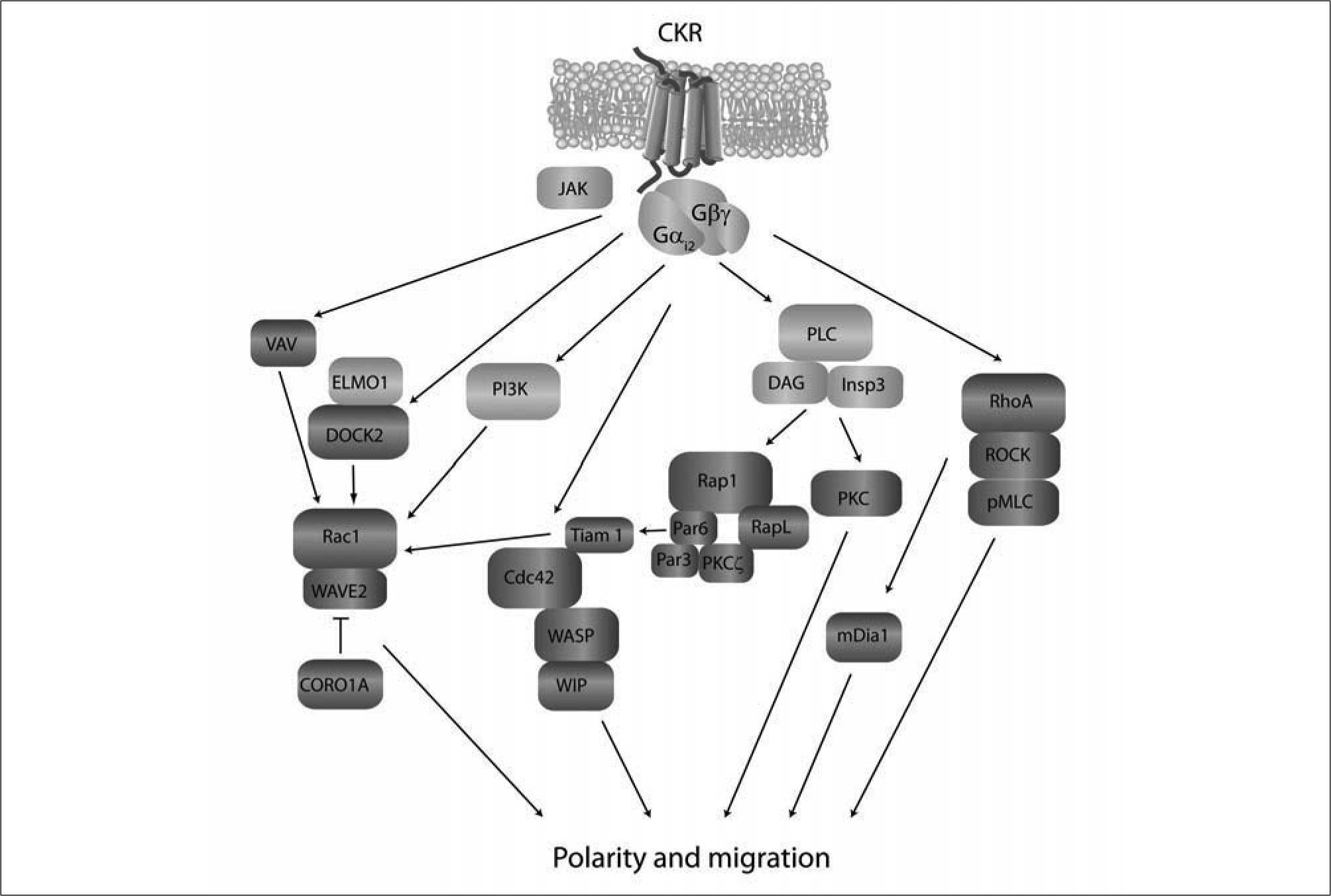
 include Gαi proteins, Jak kinases, PI3K and PLC. The exact connections and order in which these mediators are recruited to the signaling cascade is incompletely understood. These \"early\" activation signals give rise to events related with small GTPases, their activators and effectors (dark grey), which control cytoskeletal remodeling. DOCK2 is central for chemokineand S1P-mediated Rac activation which regulates polymerization of actin at the leading edge via WAVE2. This is antagonized by CORO1A, which is nevertheless required for organized polymerization/depolymerization and leading edge formation. Cdc42 regulates actin cytoskeleton via WASP and WIP and is thought to participate in stabilization of a well-oriented leading edge in response to chemokines. By interacting with RapL and Par proteins, Rap may drive activation of Cdc42 and Tiam-1, which in turn may lead to Rac activation and lymphocyte polarization, although the in vivo occurrence of this pathway is currently unclear. RhoA also plays important roles in lymphocyte chemotaxis through activation of the actin-nucleating protein mDia. The majority of these signaling molecules have been described to operate downstream of CKR and yet need to be confirmed for S1P-induced migration. p-MLC: phosphorylated myosin light chain.")
Schematic diagram of intracellular signaling pathways leading to lymphocyte polarization and migration. Membrane proximal signals (light grey) include Gαi proteins, Jak kinases, PI3K and PLC. The exact connections and order in which these mediators are recruited to the signaling cascade is incompletely understood. These "early" activation signals give rise to events related with small GTPases, their activators and effectors (dark grey), which control cytoskeletal remodeling. DOCK2 is central for chemokineand S1P-mediated Rac activation which regulates polymerization of actin at the leading edge via WAVE2. This is antagonized by CORO1A, which is nevertheless required for organized polymerization/depolymerization and leading edge formation. Cdc42 regulates actin cytoskeleton via WASP and WIP and is thought to participate in stabilization of a well-oriented leading edge in response to chemokines. By interacting with RapL and Par proteins, Rap may drive activation of Cdc42 and Tiam-1, which in turn may lead to Rac activation and lymphocyte polarization, although the in vivo occurrence of this pathway is currently unclear. RhoA also plays important roles in lymphocyte chemotaxis through activation of the actin-nucleating protein mDia. The majority of these signaling molecules have been described to operate downstream of CKR and yet need to be confirmed for S1P-induced migration. p-MLC: phosphorylated myosin light chain.
Termination of CKR signaling depends on the hydrolytic activity of the Gαi subunit which converts GTP to GDP. GDP-Gαi reassociates with Gβγ and terminates downstream signals. GTPase activity, and therefore duration of GPCR-triggered responses, is regulated by the Regulators of G protein Signaling (RGS) family of proteins which control the hydrolytic rate of Gαi-GTP through their GTPase activating capacity(39). Lack of RGS1 in B cells leads to enhanced chemotactic and adhesive responses to CXCL12 and CXCL13 in vitro, concomitant with increased homing to PLN and significantly augmented interstitial 3 dimensional (3D) velocities(37,40).
As described for other GPCR, signaling downstream of some CKR is initiated by formation of receptor dimers(34,41-43). Since homo- and heterodimerization has been observed only between certain CKR, such as CCR5 or CCR2, the occurrence and physiological relevance of CKR dimerization in the context of lymphocyte recirculation needs to be addressed in vivo using CCR7 and or CXCR4/CXCR5.
Early signaling events downstream of CKR include activation of Janus kinases (JAK) which were classically associated to cytokine receptor mediated pathways (34,44). Upon ligand binding, CKR undergo phosphorylation which in turn facilitates recruitment and activation of JAKs in a Gαi-independent fashion. In cell lines and hematopoietic precursors, pharmacological inhibition of JAK family members severely blocked chemokine-mediated responses(43-46). In line with these observations, JAK2 is rapidly phosphorylated in CCL21-stimulated primary naïve lymphocytes. TyrAG490, a pharmacological inhibitor of JAK kinases, impairs in vitro chemotaxis to CCL19/CCL21 as well as integrin activation inside PLN-HEV(47). Since TyrAG490 blocks other JAK family members and perhaps other tyrosine kinases, a thorough in vivo analysis of lymphocytes lacking different JAK family members is desirable.
CKR stimulation triggers activation of Phospholipase C (PLC) family members which generate two second messengers, diacylglycerol (DAG) and Inositol-1,4,5-trisphosphate (InsP3); the latter inducing transient mobilization of intracellular Ca2+ stores(48,49) (Figure 2). Blockage of all PLC isoforms with the pharmacological inhibitor U-73122 significantly decreases chemokine-dependent in vitro lymphocyte migration and adhesion as well as in vivo homing (CNA and Jens V.Stein, unpublished observations). Initial studies aiming to identify the specific PLC isoforms involved in CKR signaling focused on PLCβ isoforms, classically known to be activated through association with Gβγ-subunits of G proteins. In absence of PLCβ2, T cell migration was somewhat unexpectedly enhanced in response to chemokines. Nonetheless, in a separate study, lack of both PLCβ2/β2 resulted in a minor decrease in CXCL12- induced T cell chemotaxis despite completely blocking Ca2+ flux(50,51). Therefore, in vivo evidence is still required to clarify whether lymphocyte trafficking depends on PLC activity. Of note, T and B cells express PLCγ1 and PLCγ2 respectively, which function downstream of Ag receptors, and are activated upon CKR stimulation(52,53) (JVS and CNA, unpublished observations). We are currently investigating the potential physiological relevance of PLCγ-mediated signals downstream of CKR using genetically-deficient mouse strains.
Chemokine stimulation of lymphocytes induces transient upregulation of Phosphoinositide-3-kinase (PI3K) activity (Figure 2), which participates in membrane lipid metabolism and plays central roles in the regulation of diverse biological functions including cell migration(54-56). Class I PI3K convert phosphatidylinositol-(4,5)-bisphosphate (PIP2) into phosphatidylinositol-(3-5)-trisphosphate (PIP3) anchored at the inner leaflet of the plasma membrane. PIP3 serves as a recruitment signal for a variety of signaling molecules that bind PIP3 through their Pleckstrin-Homology domain (PH). A prominent role of PI3K-derived PIP3 production in response to chemoattractants has been extensively described in neutrophils and model organisms such as Dyctostelium(57,58). The ability of GPCR to directly activate the class IB PI3K p110γ, and its specific expression in leukocytes, made PI3Kγ a promising candidate to mediate PIP3 formation downstream of CKRs. Indeed, lack of PI3Kγ impairs efficient polarization and directional sensing in migrating neutrophils, thus compromising their ability to reach sites of inflammation in response to chemoattractants(59-63).
Unexpectedly, deficiency in PI3Kγ resulted only in a minor but detectable defect in in vitro migration and in vivo homing to SLO in T cells but not in B cells(64). When visualized in vivo, PI3Kγ−/− T cells displayed robust random motility inside PLN, although detailed analysis revealed a subtle increase in the amplitude of their turning angles when undergoing changes in direction, thereby suggesting a role for PI3Kγ in the maintenance of temporal directionality in these cells(65). In spite of being activated downstream of S1P1 in T cells, and partially contributing to T cell migration to S1P in vitro, PI3Kγ is not required for T cell egress from PLN in vivo(65).
Lymphocytes additionally express Class IA PI3K (p110α, β and δ), which have also been implicated in the regulation of cell motility(56,66,67). Stimulation of CXCR5 and CXCR4 in B cells leads to activation of PI3Kδ, which plays a minor role in B cell migration in vitro and homing in vivo, analogous to that of PI3Kγ in T cells(68). Whether lack of this isoform affects B cell interstitial motility has not been investigated to date. Of note, a recent study reported that pharmacological inhibition of all PI3K isoforms markedly reduced the migratory speed of T and B cells inside explanted PLN, which lead the authors to test the involvement of PI3Kα and β isoforms in random lymphocyte motility. Deficiency in PI3Kα reduced both T and B cell interstitial migration while PI3Kβ was required for optimal motility of B but not T cells(69). Further investigations will help to strictly define the exact contribution of each isoform to T and B lymphocyte trafficking.
Finally, the importance of PI3K activity in chemokinepromoted adhesion has also been studied. Although pharmacological inhibition of PI3K in T cells prevented in vitro adhesion to low integrin-ligand concentrations(54), it had no functional consequences in integrin activation in HEV as measured by intravital microscopy(54,64). This indicates that conformational changes of integrins are unaffected by PI3K inhibition and sufficient for firm adhesion under physiological shear.
Small GTPases, regulators of the cytoskeletonLymphocyte polarization, chemotaxis and adhesion are cellular events encompassing drastic changes in cell morphology which require dynamic remodeling of the cytoskeleton(31). Microfilaments are fundamental structures of the cellular backbone, formed through polymerization of subunits of globular actin. Formation, elongation, branching and retraction of these microfilaments determine cell shape and movement, and are spatiotemporally controlled by members of the Rho and Ras family of small GTPases (Rac, Rho, Cdc42 and Rap)(70). These proteins act as molecular switches, alternating between an active GTP-bound state and an inactive GDP-bound form. Their function is regulated by GTPase activating proteins (GAP) that enhance their intrinsic phosphatase activity, and Guanine nucleotide exchange factors (GEF) that promote their activation by exchanging GDP for GTP(70). Numerous GAP and GEF have been described for members of this family, introducing a considerable complexity and plasticity into the biological processes regulated by these proteins. Given their regulatory role in cytoskeletal dynamics, Rho GTPases and their regulators are required for migration of all cell types investigated so far, including lymphocytes.
Rac-related pathwaysRac GTPases are responsible for the formation of lamellipodia, actin-rich membrane protrusions at the leading edge of migrating cells(70). The ubiquitously expressed Rac1 and the hematopoietic cell-restricted Rac2 are expressed in lymphocytes and become rapidly activated upon chemokine stimulation(71). Due to its importance in germ line cell migration, genetic deletion of Rac1 results in embryonic lethality(72). Mice carrying a B cell-specific deletion of Rac1 had normal B cell numbers in SLO, indicating no major defects in B cell trafficking, although chemokine-induced responses were not addressed in detail(73). Rac2−/− T cells display reduced migration to CCL19 and accumulate in blood due to decreased homing to PLN(74). In addition, Rac activation has been suggested to participate in lymphocyte adhesion. Overexpression of RacV12 in T cell lines leads to increased cell spreading and adhesion(75). Consistently, dominant negative mutants of Rac, or Rac specific siRNA impair CXCL12-mediated integrin activation in cell lines and human peripheral blood lymphocytes (PBL)(76). However, in a different study, RacV12 expression in T cells did not significantly increase adhesion to integrin ligands. Therefore, the requirement for Rac activity during physiological integrin activation remains controversial.
DOCK2 is a member of the highly conserved Caenorhabditis elegans Ced-5, mammalian DOCK180, Drosophila melanogaster Myoblast city (CDM) family of proteins(77,78), and the major Rac-specific GEF promoting Rac activation upon chemokine stimulation of T and B cells (Figure 2)(64,79). Generation of DOCK2-/- mice uncovered its pivotal role in lymphocyte trafficking. Lymphocytes lacking DOCK2 display a profound defect in Rac activation, F-actin formation and in vitro migration to chemokines, correlating with a strong block in homing to SLO in vivo(79). Nevertheless, residual responses to chemokines and basal homing levels could be detected in DOCK2-/- lymphocytes, revealing the existence of a DOCK2-independent minor pathway that was found to depend on PI3Kγ activity in T cells and a different PI3K isoform in B cells(64). Analysis of integrin activation in DOCK2- deficient lymphocytes lead to two intriguing findings. First, chemokine-mediated integrin activation of DOCK2-/- T cells was normal as measured by in vitro and in vivo assays(64,80), indicating that in this cell type, CKR-triggered pathways leading to migration and integrin activation were partially independent and separable. Second, as opposed to T cells, B cell adhesion inside PLN and PP HEV was strongly reduced(64), clearly illustrating that significant differences exist in the molecular wiring of these two closely related cell types (Figure 3).
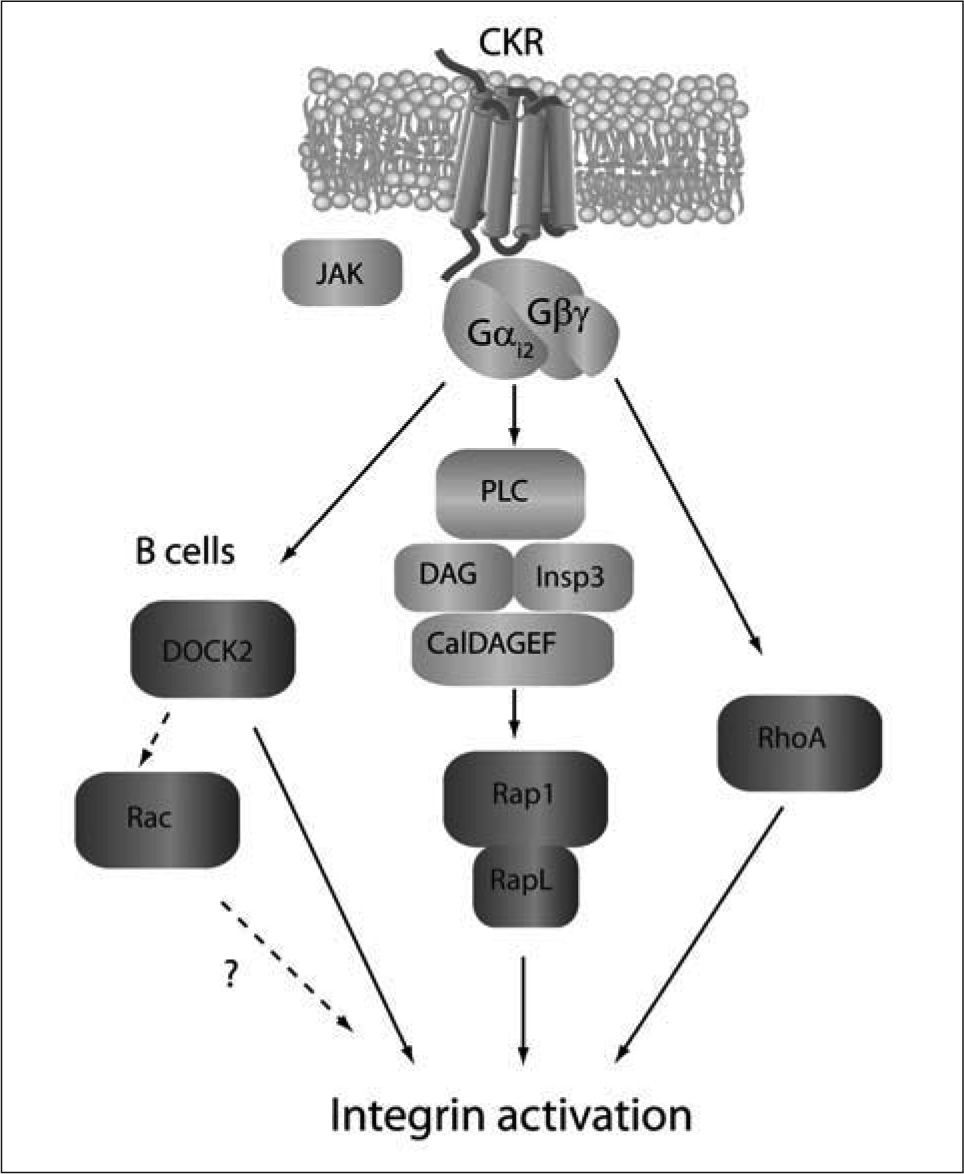
 adhesion in vivo, although it is uncertain whether this depends on its RacGEF activity or is mediated via Rac-independent mechanisms. The activators and effectors of RhoA-dependent signaling pathways required for integrin activation are yet undefined.")
Chemokine-induced signaling pathways promoting rapid integrin activation. Membrane proximal signals are depicted in light grey, while GTPaserelated signaling events are shown in dark grey. As for migration, CKR signals propagate through Gαi-mediated mechanisms. PLC-meditated activation of Rap1/RapL through CALDAGEFs is necessary to upregulate integrin adhesiveness. DOCK2 plays a major role in B cell (not T cell) adhesion in vivo, although it is uncertain whether this depends on its RacGEF activity or is mediated via Rac-independent mechanisms. The activators and effectors of RhoA-dependent signaling pathways required for integrin activation are yet undefined.
The precise mechanism by which DOCK2 deficiency leads to decreased homing in T cells has been investigated. DOCK2 is not specifically involved lymphocyte transmigration across endothelium, but affects lateral mobility of adhered lymphocytes on the apical side on the endothelium as well as of recently transmigrated T cells within the endothelial basal compartment(80). Furthermore, MP-IVM studies have demonstrated that continuous random interstitial motility of T and B cells absolutely requires DOCK2-mediated signaling. DOCK2 deficiency also results in impaired S1P1- triggered Rac activation and strongly affects the ability of lymphocytes to egress from PLN(65). Despite the crucial contribution of DOCK2 to signaling events during all steps of lymphocyte trafficking, our understanding of DOCK2- related pathways is far from being complete. In fact, apart from its known activation of Rac, DOCK2 has only two described binding partners thus far; ELMO-1 and −2 are adaptor molecules with no catalytic activity identified, which bind DOCK2 acting as necessary cooperators for its GEF activity(81). Given its large size and its crucial role in migration, it is anticipated that DOCK2 participates in numerous protein-protein interactions of potential relevance in lymphocyte trafficking, which therefore merit further investigation.
Lymphocytes express additional Rac-GEF of potential importance in CKR-induced pathways. In human PBL, CXCL12 induces Vav1 activation and rapid association with CXCR4(82). Constitutively active and dominant negative Vav1 mutants strongly attenuated CXCL12-induced polarization and migration. Nevertheless, chemotaxis to CXCL12 was not affected in Vav1−/− T and B cells, perhaps due to compensation by other Vav members(82). In T cell lines and human PBL, overexpression of dominant negative forms of Vav-1 or siRNA against Vav-1, impaired CXCL12- mediated adhesion to integrin ligands under static conditions and under flow(76). In addition, SWAP-70 is a B-cell specific Rac GEF which regulates transmigration across HEV and is therefore required for efficient B cell access to PLN. Nonetheless, chemokine-triggered Rac-GTP is not induced by SWAP-70 which rather contributes to Rac-mediated signaling events involved in transmigration of firmly adhered B cells and may therefore be chemokine-independent(83). The potential role for the Rac-specific GEF Tiam1 in chemokineelicited lymphocyte polarization will be discussed later in the context of Rap1-mediated signaling events.
Rac is suggested to promote actin polymerization via WAVE2, which in turn activates a stable complex of the actin-related proteins Arp2 and Arp3 (Arp2/3), initiating growth of new branches in actin filaments(84). Coronin1A (CORO1A) belongs to a family of actin-binding proteins which inhibit actin nucleation by interfering with Arp2/3 activity. Translocation of CORO1A to actin-rich structures of the leading edge is a key event in T cell migration(85). In vitro chemotaxis as well as homing to SLO is greatly impaired in T cells isolated from CORO1A-/- mice or a mouse strain (Ptcd mice) expressing a mutated version of CORO1A which fails to correctly localize during migration(85,86). Furthermore, interstitial motility is decreased in Ptcd T cells, which also respond poorly to S1P and therefore are defective in exiting PLN(85). Taken together, the studies mentioned thus far collectively emphasize the importance of the axis DOCK2- Rac-WAVE2-CORO1A in the signaling pathways underlying lymphocyte migration (Figure 2).
RhoAA direct assessment of the precise role of RhoA in lymphocyte trafficking has not been possible thus far due to the embryonically lethal effects of RhoA deficiency. Nonetheless, evidence from in vitro studies using lymphocytic cell lines which expressed constitutively active or dominant negative versions of this protein suggests a crucial contribution of RhoA to lymphocyte polarization and migration(87,88). Important effectors downstream of RhoA are the Rho associated coiled-coil forming protein kinases, ROCKI and ROCKII. ROCK isoforms are thought to regulate lymphocyte polarity and migration through phosphorylation of the myosin light chain (MLC) and members of the Ezrin/Radixin/Moesin (ERM) family of proteins(88,89). Pharmacological inhibition of ROCK decreases migration of human PBL to homeostatic chemokines(90,91) and prevents uropod retraction in leukocytes migrating within 3D collagen matrices(33). Genetic models are likely to further substantiate the importance of ROCK proteins in lymphocyte trafficking. Rho controls cytoskeletal remodeling through effector proteins from the mDia family of formins, which are actinnucleating proteins favoring the formation of long, straight actin filaments. Lack of mDia1 expression significantly reduces T cell homing to SLO, most likely due to its effect in migration, although integrin-mediated adhesion was not specifically measured in this study (Figure 2)(92).
In addition, a role for RhoA during chemokine-mediated integrin activation in lymphocytes has been documented. Inhibition of RhoA using C3 transferase, a Chlostridium botulinum-derived toxin, impaired chemokine-mediated adhesion of a pre-B cell line(93). Moreover, compelling evidence for the importance of RhoA was obtained when lymphocyte adhesion was measured after treatment with peptides mimicking distinct regions of RhoA, therefore blocking interaction with downstream effectors. Region-specific blocking of RhoA signaling derived in significant decrease of lymphocyte adhesion in vitro and in vivo inside PP HEV (Figure 3)(94,95).
In lymphocytes, CCL21 stimulation induces rapid activation of Protein Kinase C-ζ (PKC-ζ) which translocates to the plasma membrane in a RhoA-depedent fashion. Blocking PKC-ζ activity prevents chemokine-triggered integrin clustering and adhesion to low ICAM concentrations, similar to PI3K inhibition(94); nevertheless, the physiological relevance of these in vitro observations needs to be further investigated. Of note, the PKC family consists of at least ten members in mammalians, some of which are sensitive to activation by DAG and Ca2+ and therefore may act downstream of PLC. Although, in vitro evidence suggests that PKC activity is required for lymphocyte motility(96-98), the in vivo role of the different isoforms of this enzyme in lymphocyte trafficking needs to be thoroughly addressed by means of genetic models.
Cdc42Cdc42 is another member of the Rho GTPase family involved in the control of cytoskeletal dynamics and lymphocyte polarity(70,99,100). Since Cdc42−/− mice are not viable, its biological roles have been mainly inferred from studies using constitutively active and dominant negative mutants. Chemokines such as CXCL12 trigger rapid activation of Cdc42 in leukemic cell lines(76,101), and Cdc42 dominant negative mutants decrease migration to this chemokine(87,102). In neutrophils formation and orientation of a persistent leading edge and directional sensing are regulated by Cdc42 via its interaction with serine/threonine kinase p21-activatedkinase 1 (PAK1), and the Cdc42 and RacGEF PIXa (PAKinteractive exchange factor α(103). It was recently shown that inhibition of PAK activity and its interaction with PIX, results in defective chemotaxis to CXCL12 across membranes of reduced pore size(102). This observation is consistent with a similar role for PAK and PIX in lymphocyte and neutrophil migration; however this hypothesis has thus far not been formally addressed in vivo.
Further insight into the contribution of Cdc42 to lymphocyte migration comes from patients suffering the X-linked hereditary immunodeficiency Wiscott-Aldrich syndrome (WAS). These patients carry mutations in the gene encoding for the WAS protein (WASP) which binds active Cdc42-GTP through its Cdc42/Rac interacting domain (CRIB). Leukocytes from these patients show profound abnormalities in cytoskeletal rearrangements and chemotaxis towards CXCL12(104,105). Despite displaying only weakly reduced migration to homeostatic chemokines in vitro, WASP−/− T cells home to SLO significantly less avidly than wild type counterparts(106,107). WASP forms a heterodimeric complex with WIP (WASP-interacting protein) which contributes to the regulation of actin dynamics by stabilizing actin filaments. The deleterious effects of WIP deficiency in actin polymerization, in vitro chemotaxis and lymphocyte homing to SLO are stronger than those described for WASP−/− T cells, thus suggesting a WASP-independent role for WIP in T cell migration(106,108). Since in vitro integrin activation was normal in absence of WASP and/or WIP, the importance of these proteins maybe restricted to migration and would therefore be of interest to investigate how interfering with the WASP-WIP axis affects intranodal random motility.
RapA member of the Ras family of small GTPases, Rap1, recently gained importance for its role in integrin mediatedcell adhesion(109). Chemokine stimulation of lymphocytes induces rapid and transient Rap1-GTP formation via PLC-dependent activation of the Rap GEF, CALDAG-GEFI(110). Activation of Rap1 can be abrogated through retroviral transfer of Spa-1, a Rap-specific GAP. Spa-1 overexpression strongly reduces chemokine-promoted T cell adhesion under flow. Constitutively active Rap1V12 expression in lymphocytes mimics chemokine-mediated effects even in the absence of chemokine addition, promoting spontaneous firm adhesion(111). Taken together, these results establish a key role for Rap1 in the translation of proadhesive signals through chemokine receptors. The physiological relevance of Rap1-mediated adhesion is further highlighted by the recent discovery of a new type of human Leukocyte Adhesion Deficiency (LAD-III). Chemokine-induced Rap1-GTP formation is impaired in transformed lymphocytes derived from LAD-III patients, which correlates with defective leukocyte adhesion(110,112). In addition to its role in lymphocyte adhesion, Rap1 activity was also necessary for chemokine-triggered T cell polarization and transmigration across an endothelial monolayer(111), indicating that Rap1 regulates lymphocyte transmigration.
The relative contribution of the 4 different Rap isoforms (Rap1A, 1B, 2A, 2B) downstream of CKR remains to be fully dissected. Activation of Rap2 is required for CXCL12-mediated migration and adhesion in B cell lines(113,114). Mice bearing a lymphocyte-specific genetic deletion of Rap1A have been recently generated. Although in this study, chemokine-mediated rapid integrin activation in absence of RaplA was not measured, no obvious alterations of lymphocyte numbers of the thymus and spleen were detected in these mice(115). However, Rap1-GTP formation was still detected in Rap1A−/− lymphocytes, indicating that Rap1B activation took place. It is therefore currently unclear whether functional redundancy exists or whether other Rap isoforms play dominant roles in lymphocyte adhesion.
Some Rap interacting molecules have been described. RapL is a Rap1 binding protein, almost exclusively expressed in lymphoid tissue. RapL translocates to the leading edge of chemokine-stimulated polarized cells, where it colocalizes with the integrin LFA-1. Overexpression of RapL in T cell clones induces spontaneous polarization and upregulation of integrin adhesiveness(116). RapL−/− T and B cells showed a major block in chemokine-triggered polarization, integrin activation, and transmigration, which correlated with a strong defect in homing to SLO(117). RapL forms a complex with the serine-threonine kinase Mstl, which is thought to control lymphocyte adhesion through regulation of intracellular trafficking of integrins(118). A major unresolved question in these studies is whether and by which mechanism does Rap1-RapL-Mst1 signaling influence random motility of lymphocytes, which is known to occur in an integrinindependent manner.
Taken together, the evidence discussed so far highlights the absolute requirement for GTPase-signaling downstream of CKR and S1P1. Nonetheless, we are far from completely understanding the relative contribution, hierarchy and spatiotemporal relationships of the activation of Rac, Rho, Cdc42 and Rap1 during adhesion, polarization and directed movement of lymphocytes.
An attempt to further clarify these questions has been made studying the spontaneous polarization observed in a lymphoma line cell upon transfection of constitutively active Rap1V12. Using this model it has been suggested that activated Rap1 at the leading edge signals through Cdc42-GTP to activate membrane-recruited Par polarity complex (consisting of Par6 and Par 3) and atypical PKC-ζ. These proteins belong to the family of PDZ-containing (PSD95-Disc large-Zonula Occludens) proteins, which are known to control polarity in many cell types including lymphocytes(70,119). RaplGTP, in conjunction with Par proteins and PKC-ζ, recruit and activate Tiam1, a Rac GEF which then promotes Rac GTP loading and actin polymerization at the leading edge(120). Since CXCLl2-induced polarization and chemotaxis was only reduced by 50% in Tiam1−/− T cells, and no obvious alterations of lymphoid tissue structure are found in PKC-ζ- and Tiam1-deficient mice, the in vivo significance of this pathway needs to be further clarified.
CONCLUDING REMARKSEffective immune surveillance by lymphocytes relies on their continuous recirculation, making lymphocytes one of the most motile mammalian cell types. The main factors instructing lymphocyte recirculation through SLO have been identified. Integration of signals provided mostly by chemokines and S1P, induces T and B cells to adhere inside specific vascular beds, traverse across endothelium, crawl within tissues and access blood and lymph flow. The processing of these extracellular cues into migratory or adhesive responses requires a complex combination of receptor-mediated signals including G-proteins, membrane lipid modification enzymes, protein translocations and activation of small GTPases which ultimately control cytoskeletal remodeling.
Many of the crucial factors involved in these molecular circuits have been described, some of which have been summarized here. However, our current understanding of these signaling networks is largely incomplete and disjointed; first, many more molecular players are likely to participate in the process. Second, the knowledge acquired so far using different systems needs to be assembled in comprehensive models which manage to explain the hierarchy, kinetics, strength and spatial regulation of signaling events during migration and adhesion(32).
Experimental approaches including mass spectrometry, microarray data, novel fluorescent probes, innovative inducible genetic models, combined with in vivo subcellular microscopy, will likely provide information on the dynamics of engagement of supramolecular signaling complexes associated to migration and adhesion. Comparative analysis of the configuration, assembly and dissociation of these complexes in T and B cells but also other cell types (neutrophils, macrophages, dendritic cells) will be valuable to understand the specificity of these signaling networks and how they define certain migratory behaviors. In lymphocytes in particular, a major task will be to clarify how context-dependent encounter of chemokines results in disparate outcomes such as adhesion and migration and to separately establish the relative contribution of signaling molecules simultaneously involved in both processes (DOCK2, RhoA, PKC). Assembly of such wealth of experimental data will demand powerful bioinformatic tools to build computational models predicting and recreating signaling outcomes(121).
The precise dissection of these signaling networks will provide the basis for improved therapeutical intervention through rational design of safer, specific compounds modulating lymphocyte trafficking. The anti-α4 integrin antibody Natalizumab and FTY720, a functional antagonist of S1P1, are two drugs currently approved or under trial for treatment of multiple sclerosis. They clearly represent good examples of how exploitation of our basic knowledge on lymphocyte recirculation may prove therapeutically relevant in autoimmune diseases, transplantation medicine or cancer.
DISCLOSURESThe author declares no financial conflicts of interest.
I am grateful to Jens V.Stein for his support, critical reading and detailed revision of this manuscript. I thank César Nombela Cano for helpful advice and suggestions. C.N.A is supported by a Long-term Fellowship of the Human Frontiers Science Program Organization (LT00194/2008).