


Stiff person syndrome is a rare CNS disorder characterized by progressive muscular rigidity (trunk muscles), with superimposed spasms. High titres of antibodies to glutamic acid decarboxylase (GAD-Ab) are present in more than 70 % of patients. Adult-onset cerebellar ataxia (CA) is the second most frequent disease associated with high titers of GAD-Ab, and characterized by an almost isolated cerebellar syndrome. Both syndromes are frequently associated with autoimmune type 1 diabetes (T1D). The immunogenetic basis of SPS is supported by the DQB1*0201 allele, a susceptibility allele for T1D. Several T1D autoantigens are related to proteins of the nervous system. The concordance of both neurological diseases with T1D and the presence of anti-GAD antibodies suggest a common aetiology.
El síndrome de la persona rígida (stiff person syndrome) es una enfermedad poco frecuente del sistema nervioso central de predominio axial, caracterizada por una rigidez muscular progresiva con espasmos musculares. Se desconoce su causa, pero hay evidencia de etiopatogenia autoinmunitaria. Los anticuerpos antidecarboxilasa del ácido glutámico (GAD-Ab) están presentes en el suero de más del 70 % de los pacientes. Por otro lado, la ataxia cerebelar (CA) de inicio en adultos es la segunda enfermedad más frecuente asociada a títulos elevados de GAD-Ab, y caracterizada por un síndrome cerebelar aislado. Ambos síndromes se asocian frecuentemente a diabetes mellitus tipo 1 autoinmunitaria (T1D), enfermedad que tiene como marcador serológico los anticuerpos anti-GAD. El alelo de susceptibilidad a SPS, HLA-DQB1*0201, está aumentado en pacientes diabéticos. Diversos autoantígenos relacionados con la T1D son moléculas del sistema nervioso. El conocimiento de los procesos causantes de estas enfermedades es muy limitado, pero su concordancia, los antígenos neuronales comunes y los autoanticuerpos anti-GAD sugieren una etiología común.
Stiff person syndrome (SPS) and Adult-onset cerebellar ataxia (CA) are two rare diseases of central nervous system of unknown etiology and associated with type 1 diabetes in many patients. Type 1 diabetes (T1D) is an autoimmune disease caused by the selective destruction of insulin producing beta cells by autoreactive T lymphocytes. Both SPS/CA and T1D patients share the presence of autoantibodies against glutamic acid decarboxylase (GAD-Abs) in sera. Outside the central nervous system, a high concentration of anti-GAD antibodies also recognize pancreatic beta cells and this may explain the link between SPS/CA and T1D although the common molecular mechanism between the two conditions has not been proved scientifically.
Stiff person syndrome and cerebellar ataxia associated with autoantibodies to GADSPS is a rare CNS disorder (SPS ORPHANUMBER: ORPHA3198) characterized by progressive muscular rigidity, predominantly of the trunk muscles, with superimposed spasms1. Its prevalence is estimated at about 1/1 000 000 and more than 2/3 of patients are female. Age of onset peaks around 45 and symptoms developed months or years (table 1). The disease has not been described in members of the same family and there is no known genetic predisposition. However, an association with human leukocyte antigen (HLA) type was described: 72 % of SPS patients carried the DQB1*0201 allele2. There is good evidence for a primary autoimmune aetiology. Autoantibodies against glutamic acid decarboxylase (GAD-Abs) have been detected in more than 70 % of cases3. Adult-onset cerebellar ataxia (CA) has been recently associated with the presence of high levels of anti-glutamic acid decarboxylase antibodies4-6. Most of these patients have oligoclonal bands in the cerebrospinal fluid, GAD-Ab intrathecal synthesis and late-onset T1D besides other organ specific autoimmune disorders (thyroiditis, pernicious anemia, myasthenia gravis, psoriasis) (table 1). These features are not expected if the cause was degenerative. The majority of patients described with this syndrome were women and the main cerebellar sign described is a moderate or severe gait ataxia with a mild limb ataxia. These cerebellar symptoms prevented an independent way of life6. Nystagmus was also a frequent feature of the syndrome. The co-occurrence of SPS and CA with high titre of GAD-Ab suggests an overlap of common pathogenic mechanism.
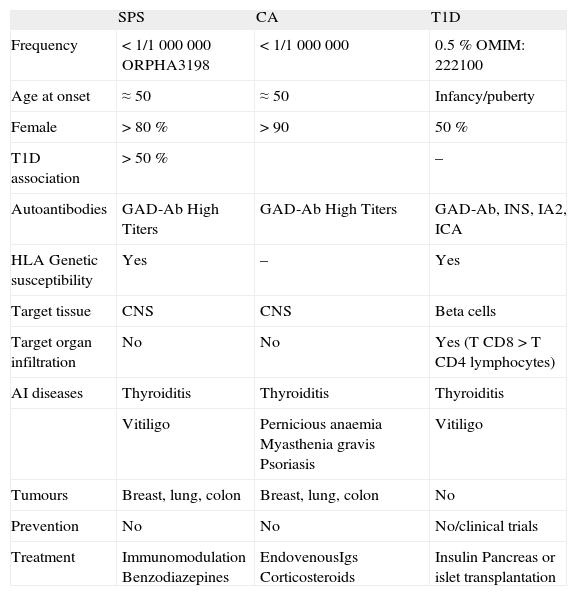
Characteristics of SPS, CA and T1D6,12
| SPS | CA | T1D | |
| Frequency | < 1/1 000 000 ORPHA3198 | < 1/1 000 000 | 0.5 % OMIM: 222100 |
| Age at onset | ≈50 | ≈50 | Infancy/puberty |
| Female | > 80 % | > 90 | 50 % |
| T1D association | > 50 % | – | |
| Autoantibodies | GAD-Ab High Titers | GAD-Ab High Titers | GAD-Ab, INS, IA2, ICA |
| HLA Genetic susceptibility | Yes | – | Yes |
| Target tissue | CNS | CNS | Beta cells |
| Target organ infiltration | No | No | Yes (T CD8 > T CD4 lymphocytes) |
| AI diseases | Thyroiditis | Thyroiditis | Thyroiditis |
| Vitiligo | Pernicious anaemia Myasthenia gravis Psoriasis | Vitiligo | |
| Tumours | Breast, lung, colon | Breast, lung, colon | No |
| Prevention | No | No | No/clinical trials |
| Treatment | Immunomodulation Benzodiazepines | EndovenousIgs Corticosteroids | Insulin Pancreas or islet transplantation |
CA: cerebellar ataxia; SPS: Stiff person syndrome; T1D: type 1 diabetes.
The syndromes are frequently associated with other autoimmune diseases6 mainly T1D (30-50 %), autoimmune thyroiditis (10 %), atrophic gastritis with pernicious anaemia (5 %), and some have tumours of the breast, lung, or colon. The two possibilities to explain the pathogenic basis of SPS are intrathecal sensitization of GAD65-reactive CD4+ T cells and synthesis of GAD65-specific autoantibodies within the CNS7 and peripheral antigen sensitization followed by CNS antigen recognition by autoantibodies that cross the blood-brain barrier. Antigen-specific CD4+ T cells are essential for the generation of high-affinity autoantibodies but there is no evidence of cellular infiltration or structural changes in the CNS of SPS patients8. However, a loss of Purkinje cells has been observed in a patient with CA associated with GAD-Ab5. This fact associated with the presence of cerebellar atrophy in magnetic resonance imaging in patients with CA and the existence of a different proliferative response to GAD with an increased production of interferon gamma suggest that despite similar humoral autoreactivity, cellular responses to GAD are different between SPS and CA9. There is evidence in vitro that GAD-Ab may block synthesis of the inhibitory neurotransmitter, gamma-aminobutyric acid (GABA) thereby attenuating inhibition of spinal motoneurones. A pathogenic role of GAD-Ab in SPS (fig. 1) is suggested by the response of symptoms to immunomodulating therapies and the correlation between antibody titres and disease outcome10.
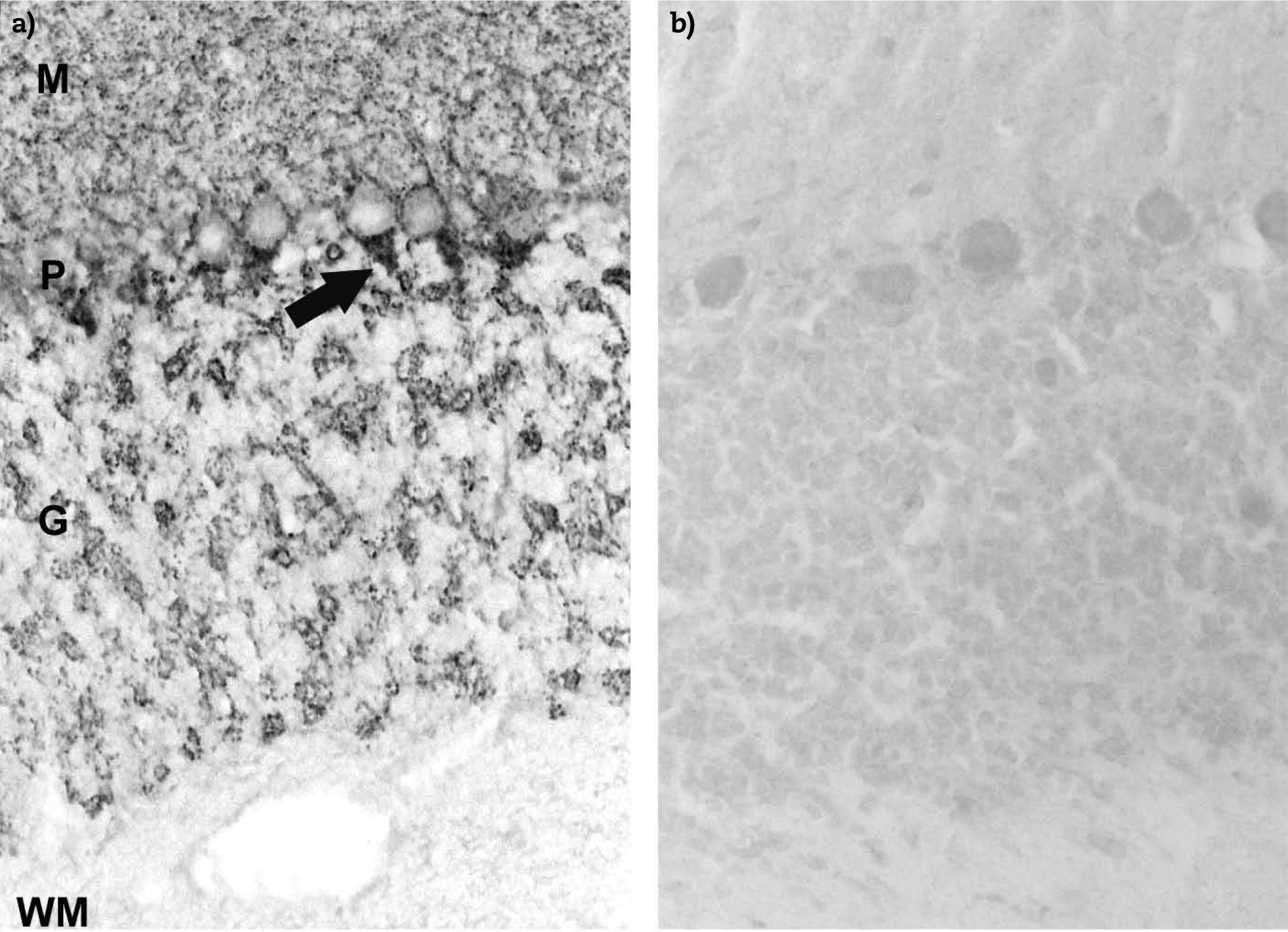
 Anti-GAD antibodies in positive sera from a SPS patient recognise the axon hillocks of Purkinje cells (P) (arrow) and diffuse nerve terminals in the molecular (M) and granular (G) layer of cerebellum. The white matter (WM) is negative; b) negative control using normal human serum.")
Immunohistochemistry staining on frozen sections of rat cerebellum. a) Anti-GAD antibodies in positive sera from a SPS patient recognise the axon hillocks of Purkinje cells (P) (arrow) and diffuse nerve terminals in the molecular (M) and granular (G) layer of cerebellum. The white matter (WM) is negative; b) negative control using normal human serum.
SPS diagnosis relies on clinical and electromyographic criteria, and is substantiated by detection of serological GAD-Ab and by characteristic electromyographic abnormalities. Benzodiazepines, baclofen and immunomodulation are standard treatments. However, the diagnosis of CA associated with GAD-Ab relies in the detection of GAD-Ab and the demonstration of a positive intrathecal synthesis of GAD-Ab. This diagnosis should be considered in female patients with T1D who develop cerebellar gait ataxia. The optimal treatment is unknown.
Type 1 diabetesT1D is an autoimmune disease of unknown aetiology caused after the selective destruction of insulin producing beta cells in the pancreatic islets11 during the infancy and the puberty (table 1). It is not considered a rare disease (OMIM: 222100) and its incidence varies depending on the geographical area12 being the highest in northern Europe and Sardinia (21 new cases per year for every 100 000 inhabitants), medium in the rest of Europe and USA (4-14 cases) and low in Japan (0,8). An annual rate of increase in incidence of 3-4 % with the largest rate of increase seen in children aged 0-4 years has been detected13.
The pathogenesis of T1D is complex and results from a combination of genetic, immunologic, hormonal and environmental factors. Genetic factors are considered a major factor in the development of the disease, because there exists a strong association between some specific alleles of HLA and other genes and susceptibility to T1D14. Although a genetic predisposition is a prerequisite for developing the disease, concordance rate in identical twins is only about 40 %, suggesting that non-genetic factors play an important role in the development of T1D. Environmental factors such as pathogens, diet, vaccines, stress and toxins also are believed to be involved in the beginning of the autoimmune process.
Several T1D autoantigens have been identified, among them insulin, GAD, carboxipeptidase H, islet-specific glucose-6-phosphatase catalytic subunit related protein (IGRP). Surprisingly, many T1D autoantigens are related to proteins of the nervous system i.e., apart of GAD, peripherin, pancreatic islet monosialo-ganglioside (GM2-1) and glial fibrillary acidic protein (GFAP) and islet-infiltrating B cells predominantly target nervous system elements15. Due to ethical and practical problems that limit access to these tissues, current understanding of the destructive processes in the diabetic pancreas was limited to morphological and immunopathological observations made in a small number of autopsies and biopsies of T1D patients16-19. These showed a leukocyte infiltration in the pancreatic islets commonly called insulitis. Thanks to improved research tools, autopsy studies of target tissue obtained from a limited number of T1D patients have, however, challenged longstanding dogmas of how the autoimmune attack develops. These studies showed that the islets from T1D have CD8+ T cells as the main subset in the insulitis, hyperexpression of HLA and adhesion molecules, proinflammatory cytokines and Fas and FasL, among other features.
Recently, the global changes at the molecular level that occur in the periphery20 and remarkably in situ in the pancreases from T1D patients21. The transcriptional profile from four T1D patients at different stages of the disease (new-onset, recent-onset and long-term) showed the complexity of the autoimmune process. Functionally altered pathways confirmed the contribution of antigen presentation genes, leukocyte subpopulations and adhesion molecules, previously identified in immunohistology studies. Remarkably, new transcriptionally altered pathways were identified i.e. chemotaxis, inflammation, innate immunity, immunoglobulin, complement, IFN-response and immunoregulation. Surprisingly, those transcriptional variations were more evident in whole pancreas than in purified islets, suggesting the involvement of the exocrine tissue. Nevertheless, the study demonstrates for the first time the presence of transcripts of immunoglobulin, innate response, antigen presentation and chemotaxis inside the islets from T1D patients. Unexpectedly, nervous system genes were significantly downregulated, a finding that supports the role of nervous system autoantigens. By contrast, regeneration genes were hyperexpressed, suggesting an attempt to restore the number of b cells. The transcriptome of the target tissue in these four cases therefore revealed a process extraordinarily more complex than previously described, highlighting the chronicity and certain heterogeneity in the autoimmune response in human T1D.
Animal models in SPSExperimental models are useful to establish the autoimmune hypothesis of the SPS. Transmission of the disease in animal models, characterization of the GAD epitope, and a search for the presence of GAD-specific T cells are strategies to be tested in animal models.
Until now, it has been demonstrated that the IgG fraction from a patient with SPS associated with amphiphysin antibodies, a paraneoplastic disorder of the CNS, induced stiffness in an animal model based on modifications of the rat experimental allergic encephalomyelitis (EAE)22. However, this model has not been probed with GAD-Ab from patients with SPS. In this sense, mice with mutation in the hypertonic (hyrt) gene exhibit severe hypertonia and have much lower levels of gamma-aminobutyric acid type A (GABA[A]) receptors in their CNS than do wild-type mice, indicating that the hypertonicity is likely to be caused by deficits in GABA-mediated motor neuron inhibition23. Because GAD-Ab catalyze the conversion of glutamic acid into GABA, the pathogenic role of GAD-Ab in neurological diseases remains a matter of debate. The administration of GAD-Ab positive sera from SPS patients in rats results in neuronal dysfunction24. Therefore, an interesting experiment may be the effect of GAD-Ab from patients with SPS and from CA in the animal model of EAE in mice susceptible to develop T1D25.
The nonobese diabetic mice (NOD), is the 'workhorse' of T1D26 and spontaneously develop a form of diabetes that resembles in some aspects human T1D. Diabetes develops in NOD mice as a result of insulitis, a leukocytic infiltrate of the pancreatic islets. Onset of diabetes is associated with a moderate glycosuria and a non-fasting hyperglycaemia. The incidence of spontaneous diabetes in the NOD mouse is 60-80 % in females and 20-30 % in males. The susceptibility to T1D is polygenic and environment including housing conditions, health status, and diet all effect development of diabetes. Genetic loci associated with susceptibility to T1D (Idd) have been identified in the NOD mouse strain. Although the main targets of the diabetogenic autoimmune attack are beta cells, nervous system that envelops pancreatic islets are also affected by the autoimmune response27.
However, few designs combining the study of both diseases have been performed in mice. A recent article demonstrates that NOD mice possessing a monoclonal GAD65-specific CD4+ T cell population develop a lethal encephalomyelitis-like disease28 in the absence of any other T cells or B cells. Autoreactive T cells were found throughout the CNS in direct concordance with GAD65 expression. In the presence of B cells, high titer anti-GAD65 autoantibodies were generated, but these had no effect on the incidence or severity of disease, suggesting that T cells alone mediate the disease. This experimental model may serve as a useful model to study GAD65-mediated diseases both of the CNS and type 1 diabetes.
SPS, CA and T1D: Cause or consequence?The link between SPS or CA and T1D is not clear. The neurological syndrome occurs in the setting of T1D or polyendocrine autoimmunity that by itself may associate with high GAD-Ab levels6,9. The concordance of the diseases, the amount of autoantigens of neural origin in T1D, and the presence of autoantibodies anti GAD suggest a possible common etiology of both diseases.
Previous studies suggested that GAD-Ab of T1D patients could be distinguished from those of SPS patients because the later recognize linear epitopes that can be identified by immunoblot29-31. However, GAD-Ab were identified by immunoblot in patients with T1D or with polyendocrine autoimmunity without associated neurological disease3,4,29,32. Presently, there are no assays that unambiguously distinguish GAD-Ab of patients with neurological syndromes.
Despite similar humoral autoreactivity, T cell responses to GAD are different between SPS and CA, with a greater inflammatory response in CA, and this difference may e relevant to the pathogenesis of these diseases9. The two possibilities to explain the pathogenic basis of SPS and CA are: a) intrathecal sensitization of GAD65-reactive CD4+ T cells and synthesis of GAD65-specific autoantibodies within the CNS, and b) peripheral antigen sensitization followed by CNS antigen recognition by autoantibodies that cross the blood-brain barrier. Until now, there is no evidence of cellular infiltration in the CNS of SPS and CA patients8. However, in a recent experimental murine model28, GAD65-specific CD4+ T cells mediate the CNS destruction and these cells were found throughout the CNS in direct concordance with GAD65 expression.
To better interpret the pathology and to advance in our understanding of the processes that lead to SPS and CA and its association with T1D it would be important to have detailed information of all the changes occurring at the molecular level. Several mechanisms of the immune response could be common to different autoimmune diseases. Overall, transcriptional alterations in those diseases affected mainly the immune system33. The neuro-immuno-endocrinological approach could lead to an innovative concept of disease pathogenesis with novel therapeutic implications.
ConclusionsIn SPS an adult-onset cerebellar ataxia associated to GAD autoantibodies (CA) the specific mechanism of disease has not been defined. Based on the evidence for an autoimmune aetiology in SPS/CA patients and on its high association with T1D and other organ-specific autoimmune diseases, common pathways of the immune response should be altered in these patients, leading to the lack of peripheral tolerance and that both diseases may have different molecular targets that cause clinical symptoms.
Understanding the pathogenesis of SPS/CA and T1D is hindered by the asymptomatic autoimmune process and by the difficulty to study the target organs directly. The finding of functional modifications in pathways of the immune response associated with SPS/CA and T1D could help to understand these two neurological diseases and its concordance with T1D and to suggest possible molecular target for therapeutic interventions.
Because SPS could be preceded by an asymptomatic period, the identification of biomarkers of progression of the disease could better identify progressing subjects and could allow immunointerventions to prevent clinical symptoms. It is also to be underlined that common diseases can correlate with rare diseases, e.g. diabetes and other autoimmune diseases with SPS, so the social interest can be considered from multiple points of view (multiple diseases). Because SPS is severe, chronic, disabling, degenerative, life-threatening, and highly painful in terms of psychosocial burden, the understanding of the cause(s) of this disease would be highly desirable for a correct diagnosis (to avoid the consequences of diagnosis delay) and for the generation of information and knowledge for the patient.
FundingOur work in this field is supported by the Spanish Ministry of Health (FIS PS09/00253). MVP is co-funded by the stabilization program of biomedical researchers of the Instituto de Salud Carlos III and Direcció d'Estrategia i Coordinació, Health Dept. of the Catalan Government.
Conflict of interestsThe authors declare no financial conflict of interests.
We thank Prof. Francesc Graus and Dr. Albert Saiz from Neurology Department, Hospital Clínic i Provincial of Barcelona for his advices and continuous support.