



Toll like receptors (TLRs) are key components of the innate immune system. TLRs signaling leads to the expression of inflammatory genes, and relies on the usage of different intracellular adaptor proteins and a tight modulation by negative regulators to attenuate inflammation. TLRs are key sensors of foreign pathogens through the recognition of pathogen-associated molecular patterns (PAMPs). In addition, emerging evidence shows that TLRs can be activated by endogenous signals released in the settings of ischemia-reperfusion, and tissue damage, which may potentially contribute to the survival or rejection of transplanted organs. This review describes the recent advances in TLR signaling and addresses the potential involvement of TLRs in organ transplantation.
Los receptores tipo Toll (TLR) son componentes fundamentales del sistema inmune innato. La activación de TLRs conduce a la expresión de genes inflamatorios y requiere de diferentes proteínas adaptadoras, así como de una regulación muy fina a través de proteínas inhibidoras que permiten el control y la finalización de la respuesta inflamatoria. Los TLRs detectan patógenos exógenos gracias al reconocimiento de estructuras moleculares asociadas a los patógenos (pathogen-associated molecular patterns, PAMPs). Además, nuevos datos indican que los TLRs pueden ser activados por señales endógenas liberadas durante procesos de isquemia-reperfusión, y daño tisular. Esta activación puede tener relevancia en los fenómenos que determinan la supervivencia o el rechazo de los órganos transplantados. En esta revisión se discutirán los últimos avances en las rutas de señalización de TLRs, así como la posible implicación de los TLRs en el transplante de órganos.
The innate immune system is the first line of defense against pathogens. The innate immunity relies on the existence of a mechanism of recognition that identifies conserved molecular structures, known as pathogen associated molecular patterns (PAMPs), broadly expressed by different groups of microorganisms. These PAMPs include lipids, lipoproteins, carbohydrates and nucleic acids(1). The recognition of PAMPs is mediated by a set of germ line-encoded receptors known as pattern recognition receptors (PRRs). This recognition enables eukaryotic hosts to reliably detect a microbial infection activating a number of signaling pathways that culminate in the induction of pro-inflammatory cytokines, chemokines, etc. One of the most important and best studied families of PRRs is that formed by Toll-like receptors (TLRs). Here, the signaling events through TLRs as well as the potential implication of TLR triggering in organ transplantation will be reviewed.
TLR structure, ligands and model of activationToll-like receptors are type I membrane proteins with a common domain organization characterized by an extracellular recognition domain composed of leucine-rich repeats (LRR), which is involved directly or through accessory molecules in ligand binding, and an intracellular Toll/IL-1 receptor homology (TIR) domain that interacts with TIR domaincontaining adaptor molecules leading to downstream signaling events.
To date, 11 human and 13 mouse TLRs have been identified that recognize distinct microbial products from bacteria, viruses, protozoa and fungi(1).
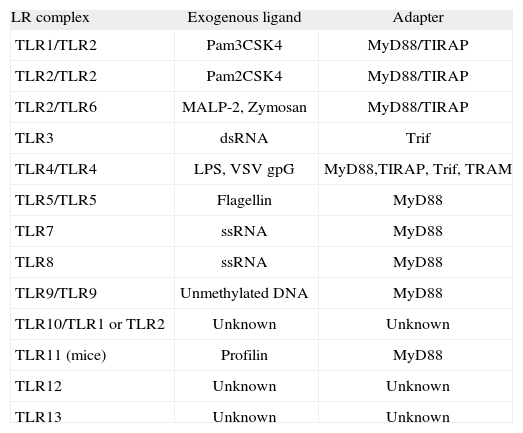
TLRs occur as dimers(2). Based on dimerization, TLRs can be classified into two categories, namely Homodimers and Heterodimers (Table I). TLR3 (which senses synthetic and viral double-stranded RNA), TLR4 (which binds Gramnegative lipopolysaccharide (LPS)), TLR5 (which detects bacterial flagellin), and TLR9 (receptor for unmethylated bacterial DNA) form homodimers. In contrast, TLR2 forms heterodimers with TLR1, TLR6, and other surface molecules such as CD36, to recognize bacterial peptidoglycan, lipopeptides, lipoproteins, mycoplasma lipopeptides and fungal zymosan. Heterodimerization facilitates discrimination between a variety of PAMPs. Thus, TLR1/TLR2 senses bacterial triacylated lipopeptides, whereas TLR2/TLR6 binds bacterial diacylated lipopeptides. TLR10 is thought to heterodimerize with TLR2 and TLR1, although a ligand for these heterodimers remains unknown(1). TLR8 (which can sense viral single-stranded RNA (ssRNA) and synthetic imidazo-quinolene compounds) has recently been shown to dimerize with TLR7 and TLR9. Interestingly, TLR8 activation antagonizes signaling by TLR7 or TLR9. Moreover, adding complexity to this scenario, TLR9 has also been shown to interact with TLR7, modulating signaling through TLR7(3). In addition, the recognition of endogenous ligands by TLRs is now thought to have an important role in the regulation of inflammation, both in infectious and noninfectious diseases. A number of endogenous ligands have been identified, including heat shock protein (HSP) 60, HSP 70(4,5), heparan sulfate(6), hyaluronan(7), fibronectin extra domain A(8), uric acid(9), oxidized LDL(10), intracellular components of fragmented cells(11,12), myeloid-related proteins-8 and 14(13), eosinophil-derived neurotoxin(14), and human defensin-3(15). As these ligands are accessible to TLRs in the setting of injury or non-infectious threat, they have been called "danger signals". However, the precise identification of these endogenous ligands has remained controversial because of their possible contamination with microbial products(16).
TLR: complex, ligands and adapter proteins
| LR complex | Exogenous ligand | Adapter |
| TLR1/TLR2 | Pam3CSK4 | MyD88/TIRAP |
| TLR2/TLR2 | Pam2CSK4 | MyD88/TIRAP |
| TLR2/TLR6 | MALP-2, Zymosan | MyD88/TIRAP |
| TLR3 | dsRNA | Trif |
| TLR4/TLR4 | LPS, VSV gpG | MyD88,TIRAP, Trif, TRAM |
| TLR5/TLR5 | Flagellin | MyD88 |
| TLR7 | ssRNA | MyD88 |
| TLR8 | ssRNA | MyD88 |
| TLR9/TLR9 | Unmethylated DNA | MyD88 |
| TLR10/TLR1 or TLR2 | Unknown | Unknown |
| TLR11 (mice) | Profilin | MyD88 |
| TLR12 | Unknown | Unknown |
| TLR13 | Unknown | Unknown |
Modified from Hoebe et al.(25).
Although the microbial ligands that activate TLRs are well known, the mechanisms of TLR-mediated recognition have remained unknown as crystallographic analysis of TLRs has been unsuccessful. A variety of LRR domaincontaining proteins have been crystallized, revealing that these domains form horseshoe-like structures with a concave face and a convex surface. Interestingly, the crystal structure of the TLR4 extracellular domain in complex with MD-2 bound to eritoran, an antagonist of LPS(17) has recently been revealed, as well as that of the TLR1/TLR2 extracellular domains complex bound to the synthetic lipopeptide agonist Pam3CSK4(18). These studies have shown that two modes of ligand recognition by TLRs exist (Fig. 1). Thus, TLR4 binds to MD-2-eritoran at the concave face of the LRR, whereas binding of Pam3CSK4 occurs at the convex surface of the LRR. Kim et al. suggested a model in which LPS interaction with TLR4/MD-2 results in allosteric changes in MD-2 that promote its binding to a second TLR4. This suggests that TLR agonists will induce TLR dimerization, whereas antagonists are likely to interfere with dimerization. One possibility suggested by these studies is that protein ligands may bind their respective TLRs as MD-2, through the concave surface of the TLR ectodomains. On the other hand, the nonprotein TLR ligands, are likely to interact with the convex surface, similarly to Pam3CSK4(17). Once ligand binding happens and conformational changes occur, the two TIR domains on the cytosolic face of each TLR get into closer proximity, creating a platform that is necessary for adaptor recruitment and downstream signaling.
.")
Models of ligand-induced TLR activation. Binding of the TLR2 ligand Pam3CSK4 occurs at the convex face of the extracellular domain, inducing TLR2-TLR1 heterodimerization. One single molecule of Pam3CSK4 is enough to induce heterodimerization. In contrast, binding of LPS to the TLR4-MD2 complex occurs at the concave face of TLR4 extracellular domain, leading to TLR4 homodimerization. Induction of homodimerization requires two molecules of both LPS and MD2 bound to each TLR4 (modified from reference 17).
In order to have a functional TLR, other proteins, in addition to the receptor itself, may be required as "coreceptors". For instance, CD14 and MD-2 are essential to obtain a complete, active TLR4 complex. CD14 is strictly required for "smooth" LPS isoforms, but not for lipid A, to elicit a signal(19). CD14 and CD36 act as co-receptors for the TLR2/TLR6 heterodimer. CD36 permits full activation of TLR2/TLR6 by diacylated lipopeptides such as MALP-2. CD36, however, is not required for TLR2/TLR1 activation, which detects triacylated lipopeptides (20). CD36 is believed to bind endogenous molecules such as oxidized LDL, which may, therefore, elicit inflammatory signals via the TLR2/TLR6 pathway(21). CD14 is required but not essential for TLR2 signaling, as CD14 deficiency reduces bacterial lipopeptide signaling but does not abolish it(19,22).
TLR signaling cascadeTLR activation triggers intracellular signaling pathways that lead to the induction of inflammatory cytokines, type I interferons (IFNs), and upregulation of co-stimulatory molecules leading to the activation of the adaptive immune response. Ligand recognition results in the recruitment of intracellular TIR-domain-containing adaptor proteins, including myeloid-differentiation primary-response protein 88 (MyD88, shared by all TLRs, except TLR3), and Toll/IL-1R domain-containing adaptor-inducing IFN-β (Trif, employed by TLR3 and TLR4)(23). Recruitment of MyD88 leads to the activation of the MAP kinases ERK, JNK and p38, and nuclear translocation of the transcription factor NF-κB, key regulators of many inflammatory response pathways (MyD88-dependent pathway)(24,25). The activation of these signaling pathways is absent in MyD88-deficient mice in response to TLRs, except TLR4 and TLR3. This is due to the activation of a second, alternative pathway triggered by Trif (Trifdependent pathway) that culminates in the activation of nuclear factor-κB (NF-κB), mitogen-activated protein (MAP)-kinases (MAPKs) and the transcription factors interferon-responsive factors (IRFs), whose are responsible for induction of type I IFNs, in particular IFN-β(26,27). Besides MyD88 and Trif, two other adaptor proteins have been described: TIR-domain-containing adaptor protein (TIRAP, required for MyD88-dependent signaling by TLR2 and TLR4), and Trif-related adaptor molecule (TRAM, required for Trif-dependent signaling through TLR4, but not TLR3)(23,28). Specific adaptor use by different TLRs combined with cell type-specific signaling pathways determine differential responses: inflammatory response, cell differentiation, proliferation, apoptosis, etc. It is worth mentioning that simultaneous activation of TLRs leads to cooperation in cellular and immune responses. Thus, simultaneous stimulation of TLR2 and TLR4 resulted in a synergistic induction of tumornecrosis factor-alpha (TNF-α) production(29,30). In addition, stimulation of macrophages with both TLR3 and TLR9 ligands showed a synergistic effect on TNF and IL-6(31). Cytokine production can also be negatively regulated by simultaneous signaling through certain TLRs(32).
MyD88-dependent pathway. Engagement of TLRs and MyD88 activates a signaling cascade including IL-1R-associated kinases (IRAKs, in particular IRAK4, which is indispensable for activation of MyD88-dependent pathway), (TNF)-receptor-associated factor 6 (TRAF6), transforming growth factor-β (TGF-β)-activated kinase (TAK1), IκB kinases (IKK) complex and the MAPK family (ERK, JNK and p38). The phosphorylation by IKK of the Inhibitor of NF-κB (IκB) is necessary for the degradation of IκB and the nuclear translocation of NF-κB, which will trigger an inflammatory gene expression program. Activation of MAPKs leads to the activation of the transcription factor AP-1 and induction of inflammatory mediators. Furthermore, activation of distinct MAPKs might determine the nature and magnitude of TLR-mediated inflammatory responses(33).
Trif-dependent pathway. The observation that TLR2, 5, 7 and 9 activation does not lead to activation of NF-κB and MAPK signaling pathways in MyD88-deficient mice, whereas TLR3 and TLR4 activation appears to activate NF-κB and MAPK, although with delayed kinetics in the latter one(34), strongly suggested the presence of a MyD88-independent pathway. Trif was identified as an essential adapter of the MyD88-independent pathway(35,36). Trif-deficient mice show defective IRF3 activation and IFN-β induction after TLR3 and TLR4 stimulation. Moreover, late phase activation of NF-κB and MAPK is totally dependent on Trif signaling(37,38). TRAF6 is essential in Trif-dependent NF-κB activation(39). In addition, receptor-interactin protein 1 (RIP1)-deficient cells show impaired activation of NF-κB upon TLR3 and TLR4 activation, indicating that RIP1 participates in Trif-dependent NF-κB activation(40).
An interesting aspect regarding TLR signaling relates to the timing of signals from TLR4. In MyD88-deficient cells, the time course of activation of NF-κB is delayed upon TLR4 stimulation, as NF-κB nuclear translocation occurs in two waves in response to LPS(41,42), namely a first wave, through engagement of MyD88 leading to IKK activation and NF-κB translocation; and then a delayed wave, through autocrine TNF-α production, which is initiated by Trif activation of IRF3.
Negative regulators. TLR signaling must be tightly controlled, as there is abundant evidence that unrestricted TLR activation may lead to acute or chronic inflammatory diseases(43). Therefore, a spatio-temporal regulation of TLR signaling is essential for limiting inflammation. A number of inhibitory molecules shut down TLR signaling by different mechanisms, including downregulation of TLR expression, sequestration of signaling molecules, blockade of their recruitment, degradation of target proteins, and inhibition of transcription, among others. Interestingly, most of these inhibitors are induced upon TLR triggering, indicating an inhibitory feedback mechanism of TLR-mediated immunity.
For instance, CD180 (RP105) blocks TLR4 signaling via sequestration of MD1(44). ST2L sequesters MyD88 and TIRAP(45). SIGIRR interacts with IRAKs and TRAF6 to block TLR signaling(46). Triad3 overexpression promotes downregulation of TLR4 and 9 via a proteosome-dependent manner(47). Deubiquitinating enzyme A (DUBA) can also inhibit TLR signaling through the efficient deubiquitination of TRAF3(48). IRAK-M, which is inducible by LPS, prevents dissociation of IRAK4 and IRAK1 from MyD88(49). Moreover, splicing variants of IRAKs and MyD88 have been isolated and implicated in sequestration of key signaling molecules(50). (IRAK)-1 binding protein 1(IRAK1BP1) downregulates TLR2- mediated IL6 production, and is a critical factor in preventing dangerous overproduction of proinflammatory cytokines by the innate immune system(51). TRAF1, when cleaved by caspase 8, binds Trif and inhibits Trif-dependent signaling(52). TRAF6 is a key target for TLR-signaling inhibitors. Thus, TRAF4, β-arrestins and A20 prevent TRAF6 function, therefore inhibiting downstream signaling(53-55). The IFN-inducible GTPase LRG47 has also been shown to negatively regulate TLR4 signaling(56). TGF-β interferes with TLR4 signaling as it promotes degradation of MyD88(57). Finally, S-nitrosylation of MyD88 may negatively regulate downstream signaling(58).
TLR localization and expressionTLRs are expressed by immune and non-immune cells (monocytes, macrophages, endothelial cells, etc). Two subcellular localizations have been described for TLRs: i) plasma membrane localization, which includes TLR1, 2, 4, 5 and 6; and ii) intracellular localization (such as endosomes) which includes TLR3, 7, 8 and 9. Intracellular TLRs sense viral and bacterial nucleic acids which are released after bacteria or virus degradation in late endosomes or lysosomes, allowing TLRs binding(59). It has been proposed that the spatiotemporal regulation of TLRs might be a safeguard against contacting self-ligands, which will contribute to avoid autoimmune diseases(60). Intracellular TLRs utilize MyD88 or Trif, whereas cell surface TLRs use MyD88 and TIRAP and/or TRAM as additional adapters, suggesting a link between adapter usage and TLR localization.
TLRs, TRANSPLANTATION AND REJECTIONThe importance of transplantation is increasing with the widespread use of organ and tissue transplantation for the treatment of end-stage diseases. However, one unsolved problem is graft rejection, as transplant tolerance is difficult to obtain. Understanding the immune responses underlying the events of tolerance and rejection will potentially provide with powerful tools to face the challenge of organ transplantation. Alloimmunity plays a critical role in transplantation and graft rejection. TLRs have been involved in modulation of alloimmune responses and tolerance through different mechanisms. For instance, TLRs activation on dendritic cells (DCs), monocytes and macrophages by graft transplantation might turn on the adaptive immune system through T cell maturation, which results ultimately in allograft rejection(61). In addition, it has been shown that monocytes are the primary infiltrating cell type in grafts during acute rejection, demonstrating a key contribution of innate immune cell types to allograft rejection(62). Finally, regulatory T cells (Tregs) function, which play a critical role in immune homeostasis and tolerance (depletion of Tregs leads to organ-specific autoimmunity) may be modulated by TLRs through the indirect stimulation of DC or via a direct activation of Tregs themselves which would facilitate allograft rejection(63,64).
Danger signals and ischemia/reperfusion injuryIn the setting of cellular injury and/or transplantation TLRs may recognize endogenous ligands, also known as danger signals, leading to the activation of a sterile inflammation, as it is not mounted against any pathogen or infection. Hyaluronan, which seems to be a ligand for TLR2 and TLR4 is highly accumulated in the lung of transplant recipients suffering from Bronchiolitis obliterans syndrome (BOS)(65), suggesting that persistent activation of TLRs through hyaluronan might play a role in lung rejection. HSP70 involvement in allograft rejection remains controversial, as no consistent effects have been reported(66,67). On the other hand, solid organ transplantation might be performed in the presence of exogenous ligands for TLRs(68). For instance, cardiac surgery has been found to increase the levels of circulating endotoxin; such ligand exposure is believed to frequently occur in heart transplant recipients(69). Furthermore, concomitant pulmonary infection at time of transplantation provides exposure to microbial ligands, which seems to be related to graft rejection(70).
Ischemia/reperfusion (IR) injury is a complex process whereby a disruption of blood flow and a decrease in oxygen delivery stimulate an inflammatory process leading to organ damage. IR injury is an unavoidable byproduct of transplantation as graft blood flow is interrupted for a period of time and is a common cause of organ dysfunction. IR injury can cause early organ failure, acute rejection and chronic rejection, making it a significant detriment to transplant survival(71). During IR settings, endogenous ligands from damaged/stressed cells have the capability to activate TLRs. Activation of TLRs triggers the release of proinflammatory cytokines and chemokines, along with the recruitment of macrophages, neutrophils and T cells, which results in a full-scale IR injury.
Studies using both TLR4-mutant and TLR4 deficient mice have demonstrated that liver injury in the setting of IR injury is mediated largely by TLR4(71,72), but not TLR2. TLR4 response is modulated by HO-1 and signal transducer and activator of transcription 4 (STAT4)(73). Interestingly, an early mediator of injury and inflammation in the setting of liver IR injury is the high mobility group box 1 protein (HMGB1), which acts as an endogenous ligand for TLR4(72). TLR4 seems to play a role in IR injury in other organs as well. Thus, TLR4 deficient mice showed reduced inflammation and smaller infarct sizes compared to wild type mice in a model of myocardial IR(74). On the other hand, TLR2 has been shown to be involved in cardiac remodeling after myocardial infarction(75). In renal IR injury, TLR2 plays a proinflammatory role in vivo, as manifested by reduced cytokine and chemokine production as well as reduced leukocyte infiltration in TLR2 deficient mice when compared to wild-type (wt) animals(76). It has been recently shown that TLR4 is critical to both the systemic and intragraft inflammatory responses that occur after cold IR in the setting of organ transplantation and that TLR4 signaling on both donor and recipient cells are involved(77).
TLRs and rejectionAs mentioned before, MyD88 is an adaptor protein shared by all TLRs except TLR3. Using a skin transplantation model, Goldstein and colleagues(78) documented that minor mismatched (HY-mismatched) allograft rejection does not occur in MyD88 deficient mice. In the absence of MyD88, male MyD88−/− male skin grafts transplanted to female MyD88−/− recipients survived much longer than wild type (wt) litter-mates after transplantation (100 vs 25 days, respectively). The abrogation of graft rejection in the absence of MyD88 resulted from lack of DC maturation, leading to diminished generation of anti-donor specific T cells and impaired T-helper 1 (Th1) immunity. Rejection of major MHC-mismatched allografts was studied by the same group in a model of skin and cardiac grafts in mice(79). Surprisingly, when MyD88 was absent from the recipient alone or from both recipient and donor, the allografts were rejected without significant delay compared with wt controls. Although DC function and Th1 immune responses were significantly reduced, T-helper 2 (Th2) response was unaffected. These results suggest that TLR-independent mechanisms might play a role. For instance, through the interaction of DCs with natural killer (NK) cells and NK-T cells(80). Therefore, it is possible that major MHC-mismatched transplants would lead to stronger TLR-independent immune responses.
Alternatively, allograft rejection may involve TLR-dependent, MyD88-independent signaling events, through Trif. Work by McKay and colleagues demonstrated that simultaneous deletion of MyD88 and Trif delays major MHC and minor antigen mismatch allograft rejection. The increased graft survival was attributed to a decrease in the ability of double knockout donor cells to migrate to the draining lymph nodes of the recipient, and delayed infiltration of recipient immune cells into the grafted organ(81).
The significance of TLR4 in organ transplantation has been demonstrated in multiple clinical and experimental studies. Two TLR4 polymorphisms, Asp299Gly and/or Thr399Ile, are associated with blunted responsiveness to TLR4 activation(82). Although the presence of one or both of the polymorphisms in the donor organ does not modify the rate of acute allograft rejection (as compared to recipients with neither mutation), there is a significant reduction in acute allograft rejection at six months post-transplant in recipients possessing one or both of the polymorphisms (as compared to recipients with neither mutation)(68). Kidney transplant recipients with the same TLR4 polymorphisms presented lower rate of acute rejection and reduced post-transplant atherosclerotic events over a 95-month follow-up period(83). However, it must be noted that in minor mismatch (same strain; male donor, female recipient) skin transplant models, the lack of TLR4 had no significant effect on transplant survival(78). These apparently conflicting data from animal experimental models and clinical observations have been explained by differences between human and murine immune responses, by organ-specific responses to TLR signaling, or by the influence of the chronic immune suppression used in clinical organ transplantation(68). Recently, Goldberg and colleagues reported that TLR4 induction in pancreatic beta cells is involved in beta cell death and islet allograft rejection after transplantation(84). In addition, indefinite cardiac allograft survival has been achieved by downregulation of TLR2 and TLR4 after serine protease inhibitor-1 (Serp-1) treatment. The prolonged allograft survival was attributed to the capability of Serp-1 to reduce the T cell intragraft infiltration, therefore modulating the transition from the innate to the adaptive immune response against the graft(85). On the other hand, mutations in the HSP70 gene, one of the TLR4 endogenous ligands, have also been shown to contribute to the development of acute renal allograft rejection(86). However, as double MyD88 and Trif mutations have synergistic effects on graft rejection, as well as the functional "cross-talk" among different TLRs, it is possible that multiple TLRs contribute to the allograft rejection(87).
Transplantation using anti-CD154 monoclonal antibody (mAb) has successfully induced tolerance or prolonged allograft survival in different animal models(88,89). Anti-CD154 mAb blocks the interaction between CD154 on T cells and CD40 on antigen presenting cells (APCs), inhibiting naive alloreactive CD8+ T cell activation. The mechanism of promoting long-term graft survival by CD154 targeted therapy also involves the induction of CD4+CD250+ Tregs. Thus, Zhai and colleagues established a murine cardiac transplant model in which tolerance was induced by a single dose of anti-CD154 mAb at the time of transplantation. They demonstrated that CD4+CD25+ Treg cells were responsible for maintaining unresponsiveness in this model(90). Interestingly, administration of TLR agonists during treatment with anti-CD154 mAb abrogates skin allograft survival induced by costimulation blockade. This effect relies on the inhibition of alloreactive CD8+ T cells apoptosis through a mechanism that requires TLR-induced type 1 IFN signaling pathways(91,92). In addition, Goldstein and colleagues(93) have shown that MyD88 deficiency acts synergistically with costimulatory receptor blockade in a fully allogenic murine skin transplant model. Indeed, costimulatory blockade in combination with a lack of MyD88 signaling is sufficient to increase allograft survival when both donor and recipient are deficient of MyD88. This effect seemed to be mediated by the increased suppressive activity of CD4+CD25+ Treg cells.
CONCLUDING REMARKSTLRs, through their modulation of innate and adaptive immune responses, are essential players in the battle for tolerance or rejection of transplanted organs. However, the molecular and cellular mechanisms involved remain poorly understood and represent an emerging field of research with potential therapeutic implications. Modulation of TLR signaling, through activation or blockade, in the settings of organ transplantation might lead to the development of new strategies to improve long-term survival of graft transplants.
DISCLOSURESThe authors declare no financial conflicts of interest.