




La oftalmoplejía crónica extrínseca progresiva (CPEO) es una miopatía que se caracteriza por ptosis y limitación de los movimientos extrínsecos oculares de carácter progresivo. Puede aparecer de forma esporádica o bien asociarse a otros fenotipos mitocondriales1. Las deleciones en el ADN mitocondrial (ADNmt) son una causa frecuente de CPEO2, si bien se han descrito otras causas como mutaciones puntuales en los ARN de transferencia (herencia materna)3 y deleciones múltiples (de origen nuclear o herencia mendeliana)4,5. El objetivo de nuestro trabajo ha sido describir las manifestaciones neurológicas y las deleciones del genoma mitocondrial en 2 pacientes brasileños afectados de CPEO con fibras rojas rasgadas (FRR).
Pacientes y método
Los 2 pacientes, un varón y una mujer de 32 y 28 años de edad, respectivamente, ingresaron en el Hospital Sarah de Brasilia con un cuadro clínico de ptosis palpebral y oftalmoparesia progresivas que los encuadran dentro del síndrome de CPEO. Ambos pacientes se sometieron a un examen neurológico detallado, electromiografía (EMG), análisis del líquido cefalorraquídeo (LCR), resonancia magnética de encéfalo, biopsia muscular y determinación de enzimas musculares. En el análisis del tejido muscular se emplearon diversas técnicas histoquímicas, entre ellas la tinción tricrómico de Gomori modificada y la citocromo C oxidasa para análisis de la actividad del complejo IV de la cadena respiratoria.
El análisis genético molecular de deleciones en el ADNmt se realizó en muestras de músculo por técnicas de hibridación Southern. Para ello, el ADN se digirió con la enzima de restricción Pvu II; los fragmentos obtenidos se separaron por electroforesis en gel de agarosa y se trasladaron a una membrana de nitrocelulosa. La detección del ADNmt se realizó mediante hibridación con una sonda de ADNmt marcada con digoxigenina y para la visualización de los híbridos se emplearon anticuerpos antidigoxigenina conjugados con fosfatasa alcalina, que cataliza una reacción quimioluminiscente, siguiendo las instrucciones del fabricante (Roche). La cuantificación del ADNmt delecionado se llevó a cabo a partir de la autorradiografía con un densitómetro láser XL Ultroscar LKB y el programa informático XL Scan gel. Una autorradiografía convencional muestra una banda de tamaño igual al del ADNmt normal (16.569 pares de bases) y, ante la presencia de deleciones, otras bandas de menor tamaño. Para determinar los límites de las deleciones se amplificó todo el ADNmt con la técnica de reacción en cadena de la polimerasa (PCR) larga, y se cortó con diversas enzimas de restricción para realizar un primer mapeado. Una vez localizada la región de la deleción, se amplificó esta zona utilizando oligonucleótidos flanqueantes y se secuenciaron los fragmentos amplificados. En uno de los pacientes (caso 1) la detección de deleciones se realizó mediante amplificación por PCR larga6, ya que no se disponía de suficiente ADN para la realización de la técnica de hibridación Southern.
Resultados
Caso 1
Varón de 32 años de edad, admitido en consulta de Neurología por presentar un síndrome clínico de 8 años de evolución consistente en ptosis palpebral asimétrica y diplopía. Durante ese período no observó otros síntomas tales como disfagia, hipofonía o disartria. La ptosis no fluctuaba a lo largo del día, y el paciente no presentaba síntomas de debilidad motora en reposo o intolerancia al ejercicio. El examen neurológico evidenció una ptosis palpebral bilateral asimétrica, estrabismo divergente a la izquierda, parálisis para la aducción y elevación de ambos globos oculares y parálisis de la convergencia ocular. Las pupilas eran isocóricas y normorreactivas. El estudio neuroftalmológico excluyó la presencia de retinopatía pigmentaria. La agudeza visual medida en ambos ojos fue de 20/400. Se apreció debilidad de la musculatura orbicular ocular y de los músculos frontal, orbicular de los labios y platisma a derecha. El examen motor evidenció una musculatura apendicular con trofismo, tono y fuerza musculares conservadas, en ausencia de síntomas de fatiga. Los reflejos osteotendinosos eran normales y simétricos.
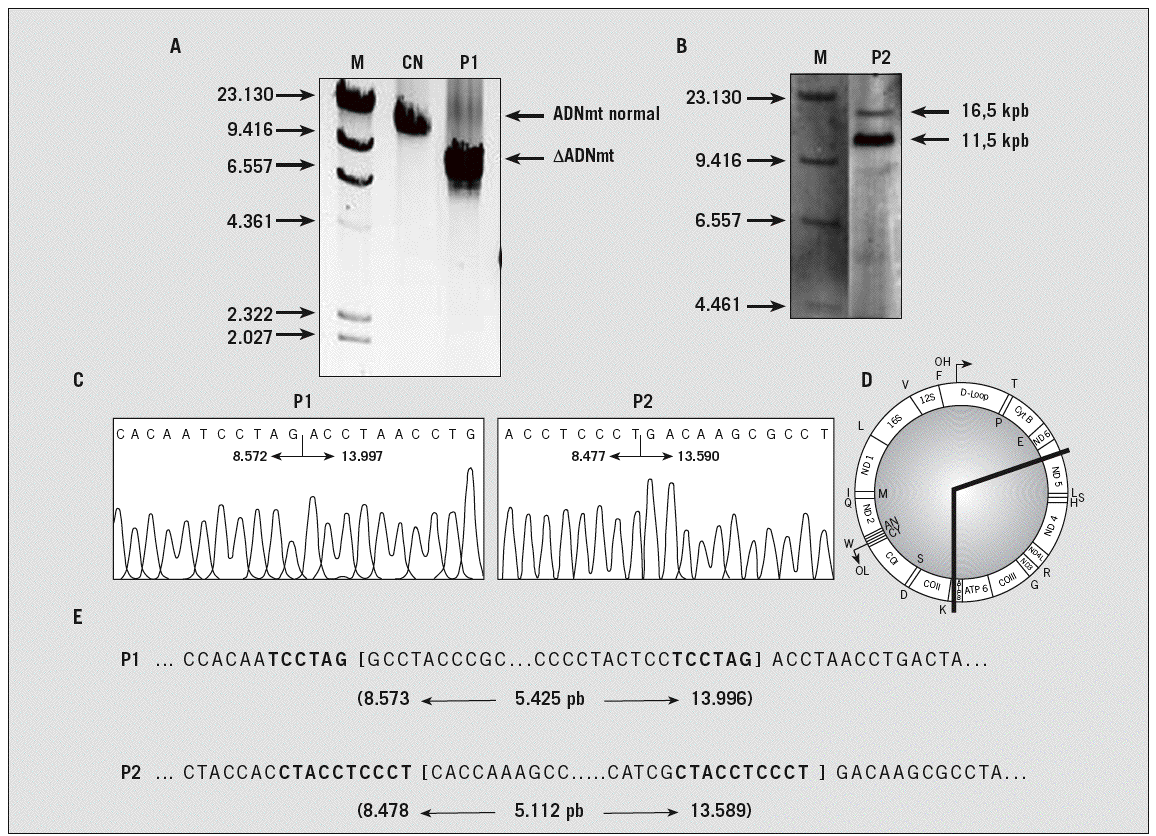
Los exámenes bioquímicos (creatincinasa: 194 U/l; valor normal [VN] < 190), amoníaco (23 µmol/l; VN: 10-47), lactato (1 mmol/l; VN: 0,67-2,47), aldolasa (3,9 U/l; VN < 7), hormona tirotropa (1,26 µU/ml; VN: 0,27-4,2), hormona tiroidea tiroxina libre (1,3 ng/dl; VN: 0,93-1,7 ng/dl), anticuerpos antimicrosómicos (15,2 U/ml; VN < 15) y antirreceptor de acetilcolina (0,01; VN: 0-0,02) fueron normales o negativos. El electrocardiograma, ecocardiograma y la resonancia magnética de encéfalo fueron normales. La EMG evidenció velocidades de conducción sensitiva y motora normales, ausencia de decremento miasteniforme en los músculos abductor menor del pulgar, trapecio, deltoides y bíceps derechos, así como en la musculatura orbicular y nasal. Una prueba con 10 mg de cloruro de edofronio intravenoso fue negativa, sin que evidenciara mejoría clínica. El estudio bioquímico del LCR fue normal (glucosa: 58 mg/dl; proteínas: 22 mg/dl; lactato: 1,2 mmol/l; 2 células, un 82% de linfocitos; índice de síntesis intratecal: 0,4, VN: 0,58-0,8). La biopsia muscular del cuádriceps derecho mostró la presencia de FRR con la tinción del tricrómico de Gomori y fibras citocromo C oxidasa (COX) negativas (figs. 1A y B). El estudio de deleciones por PCR larga evidenció una deleción de 5.425 pares de bases del genoma mitocondrial; no pudo determinarse la heteroplasmia con este método. Esta deleción estaba localizada entre las posiciones 8.572 y 13.997 del ADNmt, por lo que se pierden 11 genes (fig. 2). La deleción estaba flanqueda por una repetición directa de 6 pares de bases y no se ha descrito anteriormente. Dos años después, debido a la progresión de la oftalmoparesia, se sometió al paciente a un procedimiento quirúrgico de suspensión palpebral para tratamiento de la ptosis e injerto de fascia lata en tarso en forma de U.
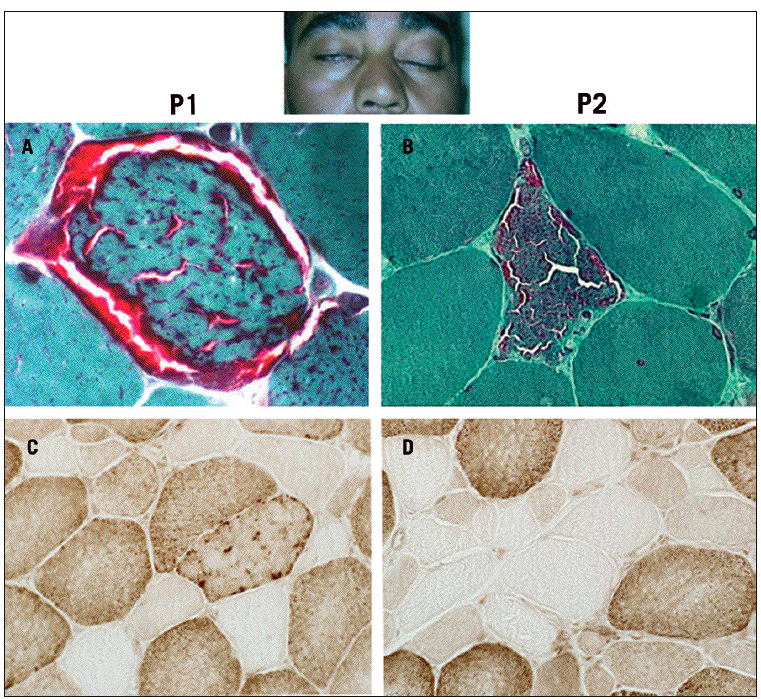
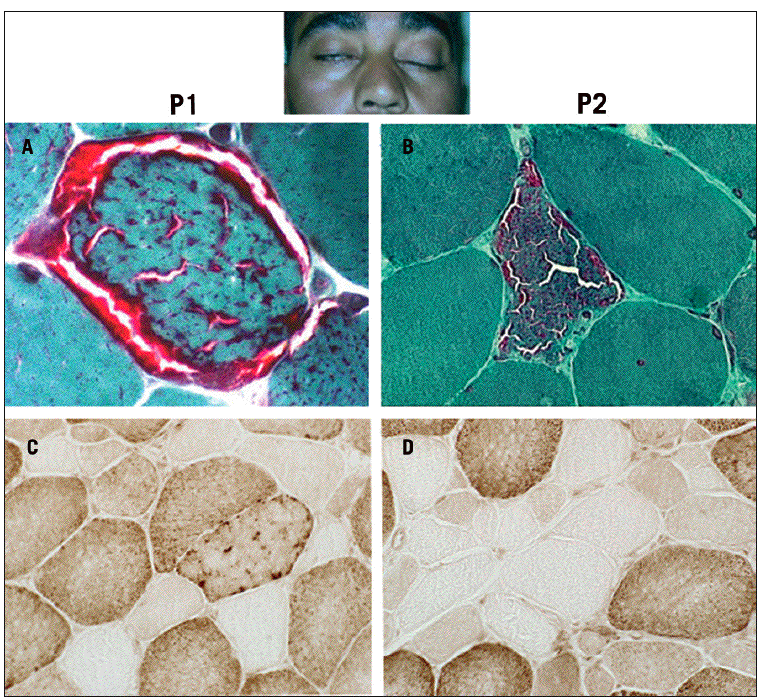
Fig. 1. Tinción del tricrómico de Gomori modificada que muestra la presencia de fibras rojas rasgadas (A y C) y actividad de la citocromo C oxidasa negativa (B y D) en la biopsia muscular de los casos 1 y 2. En la foto del caso 2 se observa la ptosis palpebral.
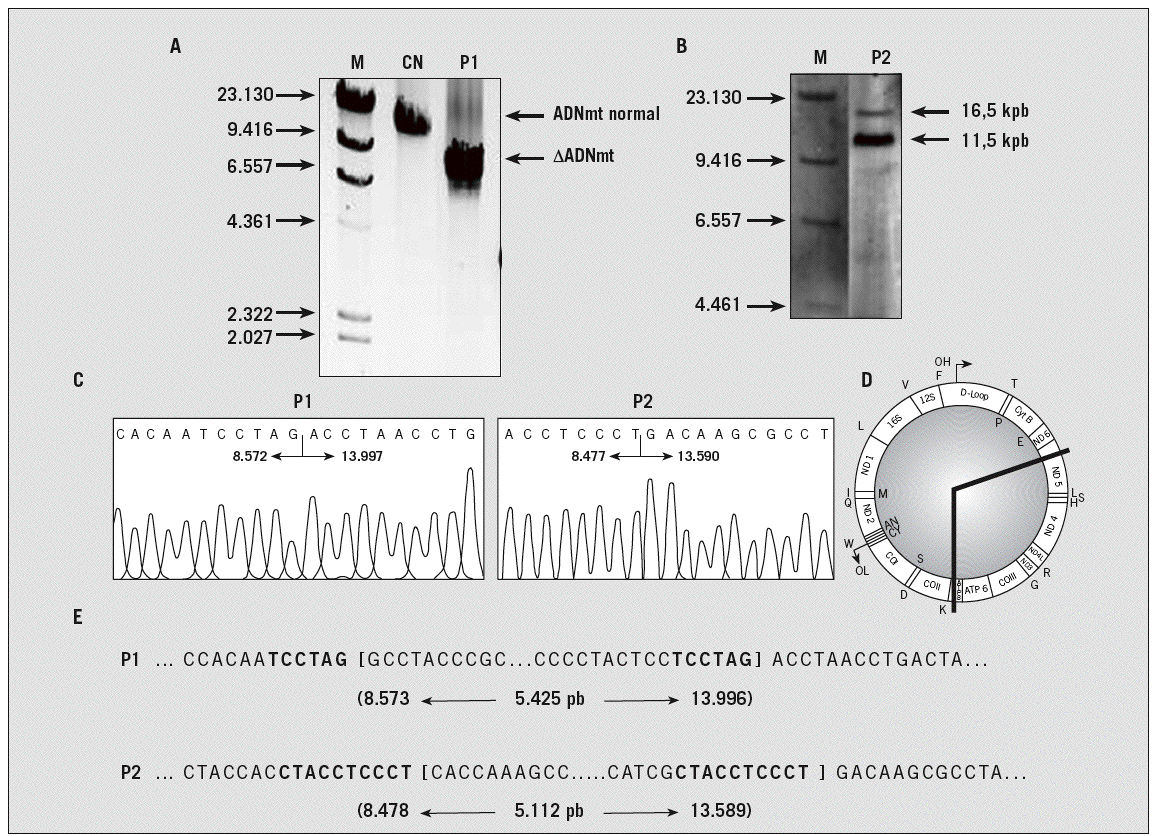
Fig. 2. Análisis de deleciones del ADN mitocondrial extraído de músculo de los casos 1 y 2 y de un control normal (CN), por técnica de reacción en cadena de la polimerasa larga (A) y por hibridación Southern (B). C: secuenciación para la determinación de los límites de la deleción que muestra el punto de rotura. D: mapa del ADN mitocondrial humano en el que se muestra la extensión de la deleción en el ADN de los pacientes. E: secuencia nucleotídica que rodea el punto de rotura de la deleción. Los paréntesis muestran los puntos de rotura. La numeración de los nucleótidos está realizada de acuerdo con la secuencia de la cadena ligera del ADN mitocondrial humano y especifica el primer nucleótido en el extremo 5' y el último en el 3' de la secuencia delecionada, así como la longitud de la deleción entre ambos. Las repeticiones directas se muestran en negrita. M: marcador de pesos moleculares, expresado en kilopares de bases (kpb); pb: pares de bases.
Caso 2
Mujer de 28 años de edad, que acudió a la consulta de Neurología por presentar un cuadro clínico de ptosis palpebral y diplopía de inicio a los 17 años de edad. Poco tiempo después de iniciados los síntomas, sintió debilidad y fatiga en los músculos proximales. Cuando contaba 19 años, se detectaron títulos discretamente elevados de anticuerpos antirreceptor de acetilcolina; ante la sospecha de miastenia gravis se inició tratamiento con prednisona y mestinón en otro centro. Durante todo ese tiempo recibió esta medicación sin ninguna mejoría, motivo por el que se la derivó de nuevo a nuestro centro para una segunda opinión. En el examen neurológico se observaron oftalmoparesia extrínseca, ptosis palpebral y exoftalmos discreto. La paciente presentaba una considerable restricción para la mirada conjugada horizontal y vertical: sólo era capaz de elevar discretamente el globo ocular en la mirada vertical; la mirada conjugada horizontal sólo era posible hacia la izquierda y estaba limitada para la derecha. La convergencia ocular estaba ausente. El reflejo fotomotor directo y consensuado estaban presentes. No se observaron señales de fatigabilidad ocular. Se observó asimismo debilidad muscular proximal contra resistencia (grados 4/5) en los músculos bíceps, deltoides y cuádriceps bilateral. El tono muscular y los reflejos osteotendinosos eran normales.
Las concentraciones de lactato en sangre (2,1 mmol/l), amoníaco (23 mmol/l), hormona tiroidea tiroxina libre (9,8 nmg), hormona tirotropa (3,42 µU/ml), iones (sodio: 141 mEq/l; potasio: 4,1 mEq/l), enzimas musculares (creatincinasa: 38 UI/l; aldolasa: 2,2 U/l) y proteína C reactiva (0,3; VN < 0,5) fueron normales, al igual que el hemograma. Los anticuerpos antirreceptor de acetilcolina se encontraban en el límite superior de la normalidad en una ocasión (0,8), mientras que en la segunda determinación fueron normales. Los anticuerpos antiendomisio mediante inmunofluorescencia indirecta fueron también negativos. El estudio bioquímico del LCR fue normal (glucosa: 56 mg/dl; proteínas: 23 mg/dl; lactato: 1,6 mmol/l; 4 células de predominio polimorfonuclear; índice de síntesis intratecal: 0,5). La EMG mostró un patrón miopático leve, con potenciales de unidad motora polifásicos, de baja amplitud y corta duración en los músculos frontal, orbicular e interóseo dorsal izquierdos, tibial anterior, iliopsoas y gastrocnemio derechos. La técnica de estimulación repetitiva mostró un discreto descenso inferior al 10% en el deltoides izquierdo. Las velocidades de conducción sensitiva y motora fueron normales en los nervios mediano, cubital y sural derechos. La resonancia magnética de encéfalo y de órbita fue normal. Una biopsia del músculo cuádriceps derecho mostró FRR con acumulación de mitocondrias en la región subsarcolémica y fibras COX negativas (figs. 1C y D). El estudio ultraestructural mediante microscopio electrónico evidenció mitocondrias morfológicamente alteradas, crestas dilatadas y estructuras paracristalinas. El análisis del genoma mitocondrial detectó una deleción de 5.112 pares de bases, con heteroplasmia del 80%, situada entre las posiciones 8.478 y 13.589 del ADNmt, que causa la pérdida de 11 genes. La deleción está flanqueada por una repetición directa de 10 pares de bases y no se ha descrito anteriormente.
Discusión
La CPEO es la forma más común de presentación de las miopatías mitocondriales. Se caracteriza por ptosis palpebral, a veces asimétrica o de inicio unilateral, y por restricción progresiva de la mirada en todas las direcciones, que se hace más evidente con la evolución de la enfermedad. La ptosis palpebral suele ser el primer síntoma y la oftalmoparesia puede cursar con diplopía, debilidad miopática proximal e intolerancia al ejercicio. La ambigüedad de los síntomas iniciales hace que esta entidad pueda confundirse con una miastenia gravis ocular, una miopatía distiroidea o una distrofia muscular oculofaríngea. La CPEO suele permanecer como un síndrome aislado de curso lento y relativamente benigno, o bien formar parte del síndrome de Kearns-Sayre7,8. En algunos pacientes, la progresiva aparición de síntomas no musculares puede empeorar el pronóstico, como es el caso de la miocardiopatía1. Las enzimas musculares pueden ser normales o bien estar discretamente elevadas. La EMG suele mostrar un patrón miopático y la biopsia muscular objetiva FRR COX negativas.
Lo llamativo de nuestros 2 casos es el inicio de los síntomas pasada la adolescencia, la dificultad para establecer el diagnóstico (la presencia de anticuerpos antirreceptor de acetilcolina falsamente positivos en el segundo paciente pudo contribuir a la demora diagnóstica) y la progresión de la oftalmoparesia con debilidad de la musculatura proximal. La presencia de FRR y fibras COX negativas en la biopsia muscular orienta al diagnóstico, pues expresan un defecto metabólico de la fosforilación oxidativa asociado a mutaciones en el ADNmt. Sin embargo, las FRR no son específicas de las miopatías mitocondriales, ya que pueden aparecer en un 1% de los pacientes ancianos sanos y, en mayor proporción, en entidades como la miopatía por cuerpos de inclusión9.
Generalmente la CPEO se debe a grandes deleciones del ADNmt que aparecen de forma esporádica y que no se transmiten a la descendencia, como sucede en nuestros 2 pacientes. Parece que el tamaño, la localización y el porcentaje de heteroplasmia de las deleciones no presentan relación con el fenotipo. Sin embargo, los 2 casos aquí descritos presentan una deleción similar, ambas flanqueadas por 2 repeticiones directas, que pueden indicar que se generaron por recombinación homóloga o por replicación errónea. Ambas deleciones, flanqueadas por repeticiones directas de 6 y 10 pares de bases, no se han descrito previamente en la bibliografía.
Los resultados obtenidos indican que esta entidad debería entrar en el diagnóstico diferencial de los síndromes de ptosis palpebral progresiva10.
