



La hiperplasia suprarrenal congénita (HSC) es una enfermedad genética causada, en la mayoría de ocasiones, por mutaciones en el gen CYP21A2. Este gen codifica la proteína 21-hidroxilasa, que participa en la conversión de la 17-hidroxiprogesterona a 11-deoxicortisol y progesterona a deoxicorticosterona, precursores del cortisol y la aldosterona, respectivamente. Por tanto, la producción de cortisol y aldosterona está alterada en los pacientes con déficit de 21-hidroxilasa.
El bloqueo en la síntesis de cortisol conduce a la estimulación de la corteza suprarrenal mediante corticotropina, y al consiguiente acumulo de precursores que se desvían hacia la síntesis de hormonas sexuales y de andrógenos suprarrenales, responsables de la virilización de los genitales externos que se observan en mujeres recién nacidas con la forma clásica de la enfermedad. Alrededor del 75% de los casos sufren, además, déficit de aldosterona con pérdida de sal por parte del riñón, lo que se traduce en hiponatremia y deshidratación grave1.
La incidencia de la enfermedad varía en función de la población a estudio, estando comprendida entre 1/10.000 y 1/20.000, y siendo responsable de entre el 90 y 95% de las hiperplasias suprarrenales2.
La enfermedad clínicamente se puede manifestar de 2 formas: la forma no clásica, que cursa con un fenotipo más leve caracterizado por hiperandrogenismo de aparición posnatal, y la forma clásica, que cursa con mayor severidad dividiéndose a su vez en 2, forma con pérdida salina y forma virilizante. La pérdida de sal, como ya se ha comentado, ocurre en al menos el 75% de los pacientes que presentan la forma clásica y lleva a síntomas de deshidratación e hipotensión en las primeras semanas de vida, que representan una causa de mortalidad neonatal importante en estos pacientes3. Las niñas con la forma clásica de la enfermedad presentan al nacer genitales ambiguos y niveles variables de virilización. Tienen un útero normal, pero con un desarrollo anómalo de la vagina. Los genitales externos en los niños son normales.
La enfermedad tiene una herencia autosómica recesiva, por lo que es necesario la presencia de 2 mutaciones, una en cada copia del gen para que se produzca la enfermedad. Se pueden, por tanto, encontrar mutaciones en homocigosis o en heterocigosis compuesta.
Existe tratamiento prenatal preventivo para evitar la virilización de los genitales externos de las niñas afectas de la forma clásica de la HSC. Debe realizarse en centros especializados con un equipo multidisciplinar de profesionales, en el que incluya el seguimiento a largo plazo del paciente. El tratamiento consiste en la administración de dexametasona a la madre en las semanas 6-8 de gestación. Dado que en estas semanas de gestación no se dispone todavía de técnicas sensibles y específicas que permitan conocer el genotipo fetal para seleccionar únicamente los embarazos de fetos femeninos afectos, este tratamiento solo está indicado en las familias que ya tienen un hijo afecto de la forma clásica pierde sal o en una pareja portadores ambos de mutación severa4.
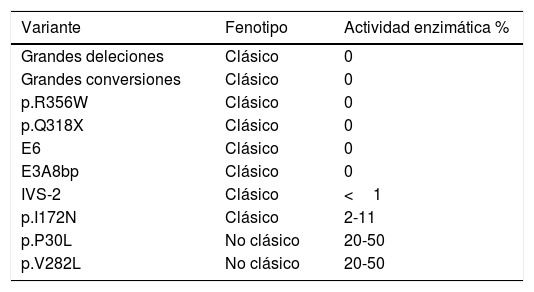
En la forma clásica, se suelen detectar 2 mutaciones graves o severas que dan lugar a una actividad enzimática prácticamente nula. En la forma no clásica el fenotipo es más leve, y se detecta al menos una mutación leve, que permite que la actividad enzimática se encuentre conservada en un 20-50%5. En la tabla 1 se muestran las mutaciones graves y leves más frecuentemente identificadas en la población española. La frecuencia de portadores de mutación en la población general es de 1/60. En algunas zonas geográficas y grupos étnicos la prevalencia es mayor debido a prácticas endogámicas6.
Variantes más frecuentemente identificadas en la población española. Se muestra el fenotipo con el que se correlacionan, así como la actividad enzimática resultante
| Variante | Fenotipo | Actividad enzimática % |
|---|---|---|
| Grandes deleciones | Clásico | 0 |
| Grandes conversiones | Clásico | 0 |
| p.R356W | Clásico | 0 |
| p.Q318X | Clásico | 0 |
| E6 | Clásico | 0 |
| E3A8bp | Clásico | 0 |
| IVS-2 | Clásico | <1 |
| p.I172N | Clásico | 2-11 |
| p.P30L | No clásico | 20-50 |
| p.V282L | No clásico | 20-50 |
En concreto, la mutación p.V282L es el cambio más frecuentemente identificado en los pacientes con HSC no clásica, se detecta en un 57% de los alelos asociados a esta forma de la enfermedad7. En combinación con otra, aunque sea de las denominadas graves, da lugar en la gran mayoría de los casos a la forma no clásica de la hiperplasia suprarrenal congénita. Recientemente, se ha descrito que menos del 3% de los pacientes que presentan esta variante y otra, ya sea leve o grave, pueden desarrollar el fenotipo clásico de la HSC. Sin embargo, el estudio genético del gen CYP21A2 es complejo y es posible que estos pacientes tengan otra mutación grave en este gen que no se haya detectado. Esto puede deberse a que normalmente no se estudia el gen completo, solo se estudian las regiones intrónicas y exónicas flanquantes8.
Cuando la mutación p.V282L se combina con otra de las denominadas graves aunque exista teóricamente este porcentaje tan bajo de desarrollar la forma clásica de la enfermedad, el tratamiento prenatal preventivo no está recomendado en estos casos dado que es todavía un tratamiento experimental y, por tanto, no está exento de riesgos. Diversos estudios en animales experimentales han demostrado que la exposición del feto a elevadas dosis de dexametasona sugieren potenciales afectos adversos sobre el desarrollo cerebral, sobre todo en las áreas ricas en receptores de glucocorticoides, como son el hipocampo y el hipotálamo, que se traducen en un anormal desarrollo motor, afectivo y en el comportamiento9.
Por tanto, dado que en parejas portadores ambos de la mutación p.V282L o de la mutación p.V282L y una mutación grave, no se instaurará ningún tratamiento ni ninguna medida preventiva para evitar tener un hijo con la forma clásica, no está recomendado el estudio de la mutación p.V282L en familiares asintomáticos de casos índices para conocer si son portadores de esta mutación10. El estudio de portadores de enfermedades genéticas autosómicas recesivas tiene sentido cuando existe alguna medida preventiva, ya sea tratamiento prenatal o diagnóstico prenatal o preimplantacional.
Por tanto, el estudio del gen CYP21A2 en familiares asintomáticos solo tiene utilidad si en el caso índice se han detectado mutaciones graves en el gen CYP21A2 que puedan dar lugar en la descendencia, en combinación con otra grave, al fenotipo clásico de la hiperplasia suprarrenal congénita.
Por último, en cuanto al diagnóstico preimplantacional, señalar que es una opción reproductiva todavía no muy extendida en la HSC debido a la complejidad del locus génico implicado en esta enfermedad, en el que existe un seudogen que dificulta su análisis en los abordajes directos. Igualmente esta opción reproductiva sólo está indicado en parejas portadores ambos de mutaciones graves en el gen CYP21A2.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
