






Conferencias clínico-patológicas: la importancia del diagnóstico multidisciplinar
Más datosMujer de 18 años, natural de Barcelona, sin alergias medicamentosas conocidas ni hábitos tóxicos. Entre sus antecedentes personales destacaba una anorexia nerviosa diagnosticada 2 años antes y que había requerido ingreso en 2 ocasiones. Al momento del ingreso, se consideraba estable, con un índice de masa corporal de 18,6 kg/m2, por lo que no precisaba de tratamiento farmacológico. Adicionalmente, presentaba una gastritis crónica sin evidencia de Helicobacter pylori diagnosticada 4 meses antes (la anatomía patológica mostró gastritis crónica leve-moderada sin actividad aguda, con hiperplasia folicular polipoide, sin atrofia ni metaplasia intestinal) en tratamiento con ranitidina. La última revisión ginecológica se había realizado un año antes del cuadro actual y había presentado la última menstruación 3 días antes del inicio de la clínica.
La paciente consultó por un dolor abdominal intermitente de predominio mesogástrico y de carácter cólico de 6 días de evolución. Durante los primeros 3 días de síntomas había presentado también diarrea, con unas 5-6 deposiciones al día, sin productos patológicos y fiebre de hasta 40° C. Había recibido ambulatoriamente 3 días de tratamiento con azitromicina con mejoría de la diarrea, pero con persistencia del dolor abdominal y la fiebre. Destacaba en la historia clínica un viaje a Croacia, la semana previa.
En la exploración física las constantes eran normales salvo por la presencia de fiebre. Presentaba un abdomen blando y depresible, doloroso a la palpación en epigastrio e hipocondrio izquierdo, sin defensa ni signos de irritación peritoneal. Los ruidos peristálticos eran normales. Se palpaba una voluminosa tumoración a nivel epigástrico de consistencia no pétrea y difícil de delimitar. No se palpaban adenopatías patológicas en ningún territorio.
Los resultados de los análisis de sangre reportaron: proteína C reactiva (PCR) de 26 mg/dL, creatinina 0,81 mg/dL, filtrado glomerular estimado por fórmula CDK-EPI de > 90 mL/min/m2, sodio de 140 mEq/l, potasio de 3,9 mEq/L, aspartato aminotransferasa (ASAT) de 23 U/L, alanin aminotransferasa (ALAT) de 12 U/L, bilirrubina total de 0,2 mg/dL con bilirrubina conjugada de 0,1 mg/dL, gamma glutamil transpeptidasa (GGT) de 37 U/L, fosfatasa alcalina (FA) de 77 U/L, lactato deshidrogenasa (LDH) de 211 U/L, amilasa de 58 U/L, lipasa de 195 U/L, recuento de leucocitos de 7,86 109/L, concentración de hemoglobina de 11,3 g/dL, hematocrito de 35%, volumen corpuscular medio (VCM) de 90,2 fL, hemoglobina corpuscular media (HCM) de 29,5 pg/c, recuento de plaquetas de 388 109/L, neutrófilos 6 × 109/L, linfocitos 1,1 × 109/L, tiempo de protrombina 71%, tiempo de tromboplastina parcial activada (TTPA) 27,7 segundos, INR de 1,15. El sedimento de orina no resultó patológico.
La radiografía de tórax fue normal. En la radiografía de abdomen se observaba aire y heces en todo el marco cólico hasta la ampolla rectal, sin distensión de asas ni otros signos de oclusión, con cámara gástrica no distendida y ausencia de signos radiológicos de neumoperitoneo.
Con base en la clínica y a los hallazgos en las exploraciones complementarias, se orientó como un síndrome febril de foco abdominal asociado a una tumoración abdominal de etiología no filiada. Por ende, tras la obtención de hemocultivos y urocultivo se inició antibioterapia empírica con ceftriaxona (2 g/d) y metronidazol (500 mg/8 h).
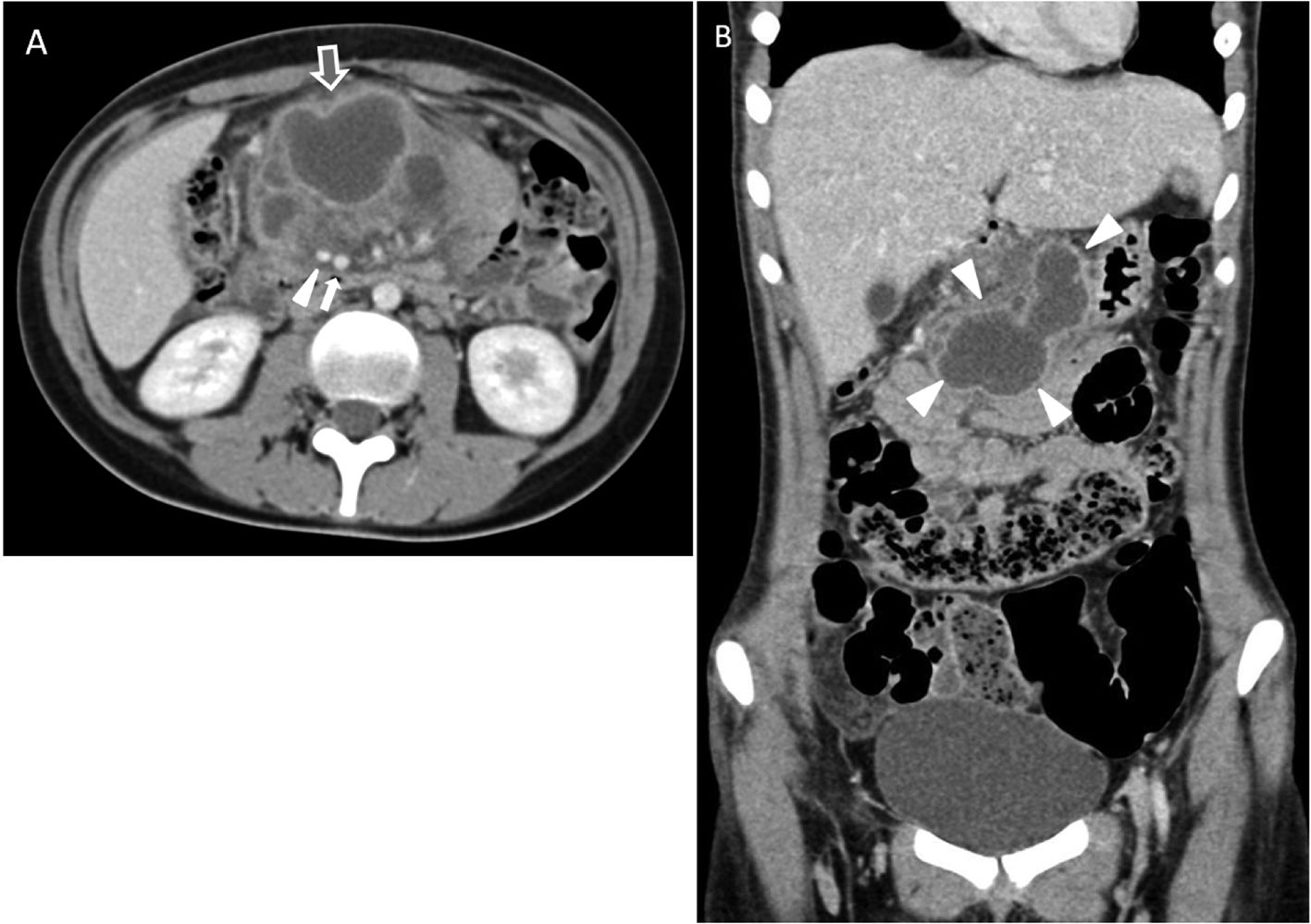
La paciente ingresó en el Servicio de Medicina Interna de nuestro centro y la tomografía computarizada (TC) abdominal evidenció una lesión retroperitoneal de partes blandas de unos 9 × 7 × 7 cm que se extendía desde el tronco celíaco hacia la arteria mesentérica superior, sin fuga de contraste oral ni intravenoso. Dicha lesión no impresionaba depender de las asas intestinales, del estómago ni del páncreas. La porción más caudal de la lesión estaba abscesificada, mostrando unas paredes hipercaptantes y lobuladas. No presentaba adenopatías abdomino-pélvicas. La imagen fue informada como sugestiva de un tumor neurogénico (paraganglioma o ganglioneuroma, entre otros) o, de forma menos probable, un linfoma (fig. 1).
 Corte axial. Se objetiva una lesión polilobulada de contenido líquido, que se extiende por el mesenterio, por delante de la arteria (triángulo blanco) y la vena mesentéricas superiores (flecha blanca), que condiciona efecto de masa, separando las estructuras vecinas (duodeno y asas intestinales). Las paredes de su borde antero-inferior muestran marcado realce y se objetiva un aumento de la trabeculación de la grasa mesentérica adyacente (flecha gris de contorno blanco), que sugiere abscesificación. B) Corte coronal. La lesión mesentérica presenta unos contornos lobulados de paredes bien definidas (enmarcada por triángulos blancos).")
TC abdominal con contraste endovenoso, adquiriéndose las imágenes en fase portal. A) Corte axial. Se objetiva una lesión polilobulada de contenido líquido, que se extiende por el mesenterio, por delante de la arteria (triángulo blanco) y la vena mesentéricas superiores (flecha blanca), que condiciona efecto de masa, separando las estructuras vecinas (duodeno y asas intestinales). Las paredes de su borde antero-inferior muestran marcado realce y se objetiva un aumento de la trabeculación de la grasa mesentérica adyacente (flecha gris de contorno blanco), que sugiere abscesificación. B) Corte coronal. La lesión mesentérica presenta unos contornos lobulados de paredes bien definidas (enmarcada por triángulos blancos).
El diagnóstico sindrómico fue el de una tumoración retroperitoneal asociada a fiebre y respuesta inflamatoria sistémica, lo que planteó un diagnóstico diferencial basado en los distintos tipos de tumores retroperitoneales.
Los tumores primarios del retroperitoneo pueden proceder de múltiples tejidos y la TC es una excelente prueba inicial para distinguirlos. En nuestro caso, por las características radiológicas, teniendo especialmente en cuenta que no dependía de las vísceras abdominales, y la edad de la paciente, se planteó como hipótesis principal un tumor neurogénico abscesificado.
Los tumores neurogénicos abdominales se desarrollan mayoritariamente en el retroperitoneo y pueden clasificarse por su origen anatómico. En primer lugar, los originados en las células ganglionares incluyen los ganglioneuromas, los ganglioneuroblastomas y los neuroblastomas; de este grupo los 2 últimos se diagnostican en su mayoría en niños menores de 5 años y son extremadamente raros en los adultos, por lo que centraremos el diagnóstico diferencial a este respecto a los ganglioneuromas. En segundo lugar, destacan los tumores originados en el sistema paraganglionar, fundamentalmente los feocromocitomas y los paragangliomas. En tercer lugar, aquellos procedentes de la vaina nerviosa, como los schwannomas, los neurofibromas y los tumores malignos de la vaina de nervio periférico (Malignant Peripheral Sheath Tumor). Las pruebas de imagen como la TC o la resonancia magnética (RM) son muy útiles en el diagnóstico diferencial, basado en el análisis de su localización, morfología y estructura interna. En general, que una masa retroperitoneal se origine cerca de la médula espinal, del músculo psoas o de los forámenes neurales sugiere un origen neurogénico.
Los ganglioneuromas son tumores neurogénicos raros y benignos procedentes de la cadena simpática paravertebral y, ocasionalmente, de la glándula adrenal. Su localización más frecuente es el retroperitoneo, en el 32-52% de los casos, seguido del mediastino posterior (39-43%) y la región cervical (8-9%). Afecta mayoritariamente a niños y adultos jóvenes. Generalmente son asintomáticos y se detectan de forma incidental. En ocasiones, pueden manifestarse en forma de dolor abdominal o de una masa abdominal palpable. Se han descrito ganglioneuromas hormonalmente activos que secretan catecolaminas, péptido intestinal vasoactivo u hormonas androgénicas, y se manifiestan con hipertensión arterial, diarrea o virilización. En la TC y la RM, los ganglioneuromas se presentan como masas bien delimitadas, ovales, en forma de medialuna o lobuladas. En la TC sin contraste, los ganglioneuromas son homogéneos y con menor densidad que el músculo. En el 20% de los casos contienen calcificaciones, que son habitualmente puntiformes. Una de sus características radiológicas es la tendencia a rodear grandes vasos, pero con poca o ninguna afectación de su luz. En la RM son también homogéneos, hipointensos en T1 y con una hiperintensidad en T2 que depende de la proporción entre la matriz mixoide y el componente celular o de las fibras de colágeno del ganglioneuroma (cuanta más matriz mixoide, mayor intensidad en T2). En la TC y en la RM con la administración de contraste pueden tener un refuerzo desde moderado hasta intenso, pero característicamente tardío, debido a que el contraste se acumula en la matriz mixoide del tumor de forma progresiva y lenta. Se distinguen en la RM por tener bandas curvilíneas de baja intensidad en T2 que corresponden a haces longitudinales y transversales de las células de Schwann y fibras de colágeno que se cruzan entre ellas en el interior del tumor1.
Como ya se ha comentado, teniendo en cuenta la edad de presentación y las características radiológicas de la TC inicial (homogeneidad, hipodensidad y afectación vascular rodeando los vasos mesentéricos), un ganglioneuroma o un paraganglioma fueron los planteamientos diagnósticos iniciales.
Los feocromocitomas y los paragangliomas son tumores neuroendocrinos procedentes del sistema nervioso autónomo. Los feocromocitomas se originan en la glándula adrenal y los paragangliomas fuera de ella, por lo que en nuestro caso se sospechó inicialmente un paraganglioma. Estos pueden clasificarse como simpáticos o parasimpáticos. Por un lado, los simpáticos tienden a ser funcionantes y sintomáticos. Los signos y síntomas son causados por una hipersecreción catecolaminérgica paroxismal y consisten en hipertensión arterial, palpitaciones, náuseas, sudación y cefalea. La clínica depende de la catecolamina producida. La mayoría de los paragangliomas funcionantes secretan noradrenalina y cursan con hipertensión arterial, pero los productores de adrenalina o dopamina presentan una clínica más variable e inespecífica, incluyendo mareo y náuseas. Por otro lado, los parasimpáticos suelen ser no funcionantes y manifestarse por efecto masa2.
En el caso de una masa retroperitoneal no anatómicamente dependiente del riñón ni de la glándula suprarrenal, es importante diferenciar los paragangliomas de otros tumores. El estudio inicial de un paraganglioma incluye la medición de metanefrinas en plasma o en orina de 24 h. No se recomienda la biopsia de la masa hasta haber descartado un paraganglioma por el alto riesgo de crisis hipertensiva y taquiarritmias3. Para evitar la fluctuación tensional perioperatoria debe realizarse previamente un bloqueo secuencial con alfabloqueadores y expansión de volumen seguido de betabloqueadoress. La resección quirúrgica es el único tratamiento curativo de los paragangliomas.
Los paragangliomas están hipervascularizados, por lo que se caracterizan en la TC por un refuerzo marcado de vasos intratumorales tras la administración del contraste. En la RM presentan una señal hipointensa en T1, pero intermedia-alta en T2; en cambio, tras la administración de gadolinio presentan un refuerzo intenso. Asimismo, son típicos los vacíos en la señal de flujo en las secuencias de eco de espín (spin-echo) dando un patrón característico en «sal y pimienta»3.
Los paragangliomas tienen un comportamiento más agresivo que sus homólogos adrenales. Aproximadamente, el 20-42% de los paragangliomas son capaces de metastatizar, comparado con el 10% de los feocromocitomas. Los sitios más comunes de metástasis son los ganglios regionales, el hueso, el hígado y los pulmones. La histología de un paraganglioma benigno y de uno maligno es la misma, por lo que se requiere un estudio de extensión para detectar malignidad, ya sea con tomografía por emisión de positrones (PET-TC) o con gammagrafía con metayodobencilguanidina (MIBG) octeótrido4.
El 40% de los paragangliomas se relacionan con síndromes genéticos como la neoplasia endocrina múltiple (MEN) 2 y MEN 3 (anteriormente conocidos como 2A y 2B), el síndrome de von Hippel Lindau, la neurofibromatosis tipo 1 (NF1), las mutaciones del complejo enzimático de succinato deshidrogenasa o el recientemente descrito síndrome de Pacak-Zhuang (paragangliomas asociados a policitemia secundaria a valores elevados de eritropoyetina). Por lo tanto, si se confirma el diagnóstico de paraganglioma se recomienda realizar un estudio genético al paciente. Se pueden evaluar todos los posibles genes implicados o bien, dado los costes de dicho estudio, priorizar según las características clínicas de acuerdo con las guías de la Sociedad Americana de Endocrinología. Si se confirma uno de los síndromes mencionados se llevará a cabo el estudio genético también a los familiares de primer grado4,5.
Esta paciente presentaba una masa retroperitoneal sugestiva de paraganglioma por TC, por lo que era prioritario descartar la producción de catecolaminas por parte del tumor de forma previa al planteamiento de una biopsia diagnóstica o una resección quirúrgica. Debe destacarse que no existía historia de hipertensión arterial, otros síntomas típicos ni antecedentes familiares en la paciente. La determinación de metanefrinas en plasma fue negativa (metanefrina 17 pg/mL para valor de normalidad < 90 pg/mL; normetanefrina 48 pg/mL para valor de normalidad < 200 pg/mL).
Los schwannomas son los tumores neurogénicos benignos retroperitoneales más frecuentes y corresponden al 6% de todos los tumores retroperitoneales primarios. Son más comunes en adultos jóvenes o de mediana edad y son el doble de frecuentes en mujeres. Suelen estar en la región paravertebral o presacra. Tienen un aspecto encapsulado con fibras que rodean el tumor. Los schwannomas tienen 2 componentes diferenciados llamados tipo Antoni A y B. El tipo Antoni A consiste en áreas con una alta densidad de células fusiformes que a menudo tienen una disposición en empalizada o de organoide. El Antoni tipo B son espacios edematosos e hipocelulares que pueden formar quistes. En la TC, el schwannoma suele visualizarse como una masa redondeada y bien delimitada que frecuentemente contiene quistes o calcificaciones. Estas calcificaciones son puntiformes, moteadas o curvilíneas a lo largo de las paredes de la masa. La captación de contraste suele ser heterogénea, por una parte, al haber zonas con mayor y menor celularidad y, por otra, debido a la formación de quistes y hemorragias. Estos mismos fenómenos hacen que los schwannomas tengan una intensidad variable en la fase T2 de la RM. En la fase T1 son hipointensos y tras la administración de contraste se refuerza la periferia del tumor y las áreas más sólidas. Estos tumores se orientan a lo largo del eje del nervio del que se originan, que puede distinguirse proximal y distalmente, y en ocasiones puede verse comprimido en la periferia del tumor1,6.
Los neurofibromas son en el 90% de los casos tumores solitarios y el resto corresponden a casos de NF1 (si bien el 30% de los pacientes con neurofibromas aislados padecen una NF1). Se diagnostican entre los 20 y los 40 años y son más frecuentes en varones. Al contrario que los schwannomas, no están encapsulados ni tienen componentes diferenciados. Tampoco desplazan los elementos fasciculares del nervio de origen, sino que interfieren con ellos. Están constituidos por una proliferación difusa de células de la vaina del nervio entrelazadas con gruesas fibras de colágeno más centrales y grados variables de degeneración mixoide que pueden formar múltiples quistes más bien en la periferia del tumor. Esta disposición les puede dar un aspecto de diana característico. En la TC son homogéneos, redondos y de densidad similar al músculo. Captan poco contraste, pero de forma homogénea, en parte debido al importante componente de fibras colágenas. El tumor maligno de la vaina periférica corresponde principalmente a la malignización de un neurofibroma y la mitad de los casos se dan en pacientes con NF1. Clínicamente, se manifiesta como dolor neuropático, déficit motores o sensitivos1,6,7.
Un segundo bloque de enfermedades que debían incluirse en el diagnóstico diferencial inicial eran los linfomas, aunque la imagen radiológica era menos sugestiva de esta entidad. Los linfomas constituyen un tercio de las neoplasias retroperitoneales primarias. Por la edad de la paciente, hubiera sido más probable un linfoma de Hodgkin, cuyo diagnóstico tiene una distribución bimodal en la tercera y la séptima décadas de la vida. Aunque la mayor parte de los linfomas se presentan en forma de adenopatías patológicas en múltiples territorios, ocasionalmente pueden presentarse en las pruebas de imagen como masas únicas bien definidas y homogéneas, con un refuerzo moderado que se extiende entre las otras estructuras sin comprimirlas8.
Con intención de caracterizar mejor la lesión retroperitoneal y, teniendo en cuenta que el síndrome febril no se justificaba de forma exclusiva por las enfermedades previamente planteadas (si bien ya se ha mencionado que la TC sugería la posibilidad de abscesificación de la tumoración), se decidió completar el estudio mediante RM.
En la RM, la lesión presentaba una morfología multiquística de aspecto arracimado, hiperintensa en T2, que podía corresponder a un linfangioma retroperitoneal gigante (8 × 5 × 9 cm) que englobaba los vasos mesentéricos y esplénicos sin infiltrarlos. Se confirmó adicionalmente una gran zona de abscesificación (7 × 5 × 2,6 cm) de morfología bilobulada (fig. 2).
. A) Secuencia potenciada en T2 en la que se observa una lesión polilobulada e hiperintesa, que se extiende por el mesenterio (enmarcada por flechas blancas). B) Secuencia potenciada en T1 con contraste endovenoso extracelular (gadolinio), donde se objetiva la lesión anteriormente descrita con paredes bien definidas e hipercaptantes. Los hallazgos muestran una lesión de contenido líquido de paredes bien delimitadas que realzan tras la administración de contraste.")
RM de hemiabdomen superior (cortes axiales). A) Secuencia potenciada en T2 en la que se observa una lesión polilobulada e hiperintesa, que se extiende por el mesenterio (enmarcada por flechas blancas). B) Secuencia potenciada en T1 con contraste endovenoso extracelular (gadolinio), donde se objetiva la lesión anteriormente descrita con paredes bien definidas e hipercaptantes. Los hallazgos muestran una lesión de contenido líquido de paredes bien delimitadas que realzan tras la administración de contraste.
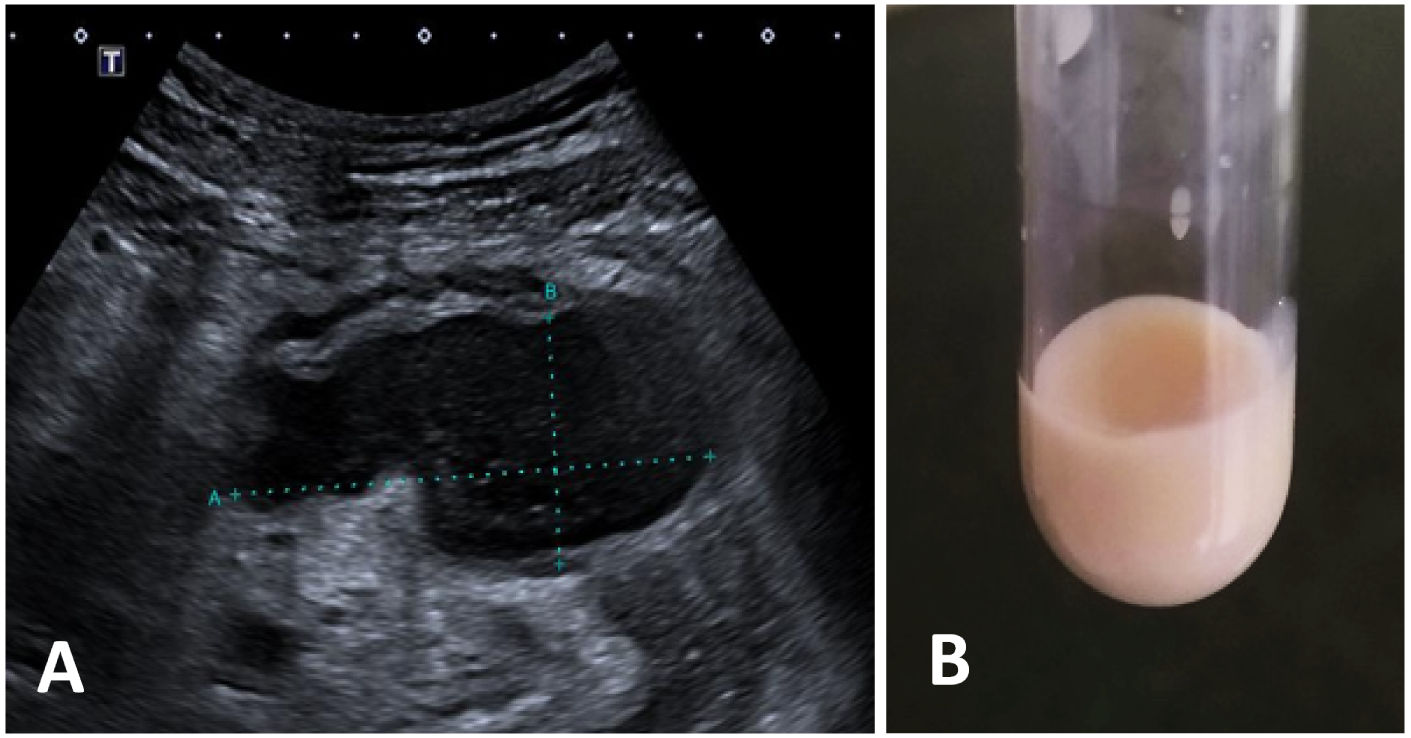
Se consideró la posibilidad de realizar una laparoscopia o laparotomía exploradoras para toma de biopsias, estudios microbiológicos, drenaje del absceso, e incluso una resección completa de la tumoración. Sin embargo, las relaciones anatómicas de la lesión implicaban un alto riesgo quirúrgico, por lo que finalmente se optó por la realización de un estudio y un drenaje de la colección mediante una punción ecoguiada (fig. 3A).
 Lesión anecoica de bordes bien definidos, que por sus características sugiere contenido líquido. Por su localización es accesible para punción aspirativa guiada por ecografía. B) Líquido obtenido mediante punción de la colección, del cual destaca el característico aspecto lechoso o quiloso del líquido linfático, condicionado fundamentalmente por su alto contenido lipídico.")
Ecografía y líquido obtenido mediante punción. A) Lesión anecoica de bordes bien definidos, que por sus características sugiere contenido líquido. Por su localización es accesible para punción aspirativa guiada por ecografía. B) Líquido obtenido mediante punción de la colección, del cual destaca el característico aspecto lechoso o quiloso del líquido linfático, condicionado fundamentalmente por su alto contenido lipídico.
La punción de la lesión condujo a la obtención de un líquido de aspecto quiloso (fig. 3B). El análisis del líquido mostró triglicéridos (quilomicrones) de 4.025 mg/dL, LDH de 10,435 U/L y glucosa de 1 mg/dL. La citología del material drenado informó de extensiones de fondo inflamatorio con abundantes neutrófilos y detritus. No se observaron atipias celulares.
La abundante presencia de quilomicrones en el líquido drenado confirmó el origen linfático de la lesión que sugería la RM.
De la muestra obtenida se cursaron también cultivos microbiológicos y dada la presión antibiótica que la paciente estaba recibiendo, se decidió también realizar un estudio por reacción en cadena de la polimerasa (PCR) basado en la detección de rRNA 16S. Tanto la detección génica como el cultivo permitieron identificar colonias de Salmonella typhimurium (S. typhimurium) como responsable de la abscesificación de la tumoración.
Tras el aislamiento microbiológico, se modificó el esquema antibiótico a cefotaxima y ciprofloxacino de forma consensuada con el Servicio de Enfermedades Infecciosas.
Diagnóstico clínicoTumoración quística retroperitoneal abscesificada. Probable linfangioma.
DiscusiónLa punción del contenido de la colección, que obtuvo líquido linfático, permitió caracterizar la tumoración como de origen linfático, confirmando así el diagnóstico sospechado mediante RM.
Los linfangiomas quísticos son tumores benignos que consisten en una malformación rara del sistema linfático que acontecen en uno de cada 4.000 nacidos vivos9. Cerca del 90% se diagnostican antes de los 2 años. El 90% ocurren en la cabeza, el cuello y las axilas, mientras que un 5% se dan en el mesenterio, las vísceras abdominales, el retroperitoneo, el pulmón o el mediastino. Sin embargo, son los tumores más frecuentes del mesenterio10. Los linfangiomas retroperitoneales suponen el 1% de todos los linfangiomas, causados por un fallo de comunicación entre el tejido retroperitoneal linfático y los vasos linfáticos principales. Se han reportado aproximadamente 300 casos hasta la fecha. Son más frecuentemente detectados en la edad adulta, posiblemente debido a que no causan defectos estéticos como los de cuello y la axila. Clínicamente, pueden presentarse como una masa palpable abdominal. Habitualmente, plantean dilemas en los diagnósticos con otros tumores quísticos retroperitoneales como aquellos originarios del hígado, el riñón o el páncreas10.
En las pruebas de imagen, los linfangiomas quísticos se caracterizan por una cavidad quística bien delimitada con una pared fina, uni o multilocular. Es característica la forma elongada y con múltiples tabiques. Muy raramente tienen calcificaciones, que son más típicas de los casos diseminados. El diagnóstico se basa en los hallazgos radiológicos, siendo la RM la prueba más confiable. La detección de gotas de grasa (material quiloso) dentro del quiste confirma el diagnóstico de linfangioma. En caso de obtenerse una muestra para estudio histológico, la anatomía patológica consiste en vasos linfáticos de diferentes tamaños rodeados de linfocitos6,11.
Estos tumores crecen progresivamente hasta comprimir las estructuras adyacentes. El crecimiento súbito de linfangiomas suele ser debido a sobreinfección o a una hemorragia intraquística9. En el caso que presentamos, el crecimiento del tumor se debía en gran parte a una sobreinfección y disminuyó de forma significativa tras el inicio del tratamiento antibiótico.
El tratamiento de este tumor benigno está condicionado por su localización y su extensión. Los linfangiomas quísticos pueden rodear y encapsular a otros órganos haciendo muy compleja su resección quirúrgica, requiriendo una valoración multidisciplinar12. En los casos en que se desestima la opción quirúrgica, se ha demostrado la efectividad y la seguridad de la escleroterapia con diversas sustancias, así como el drenaje de la lesión13,14.
En el caso de esta paciente, el compromiso de la arteria y la vena mesentéricas superiores condicionaba una elevada complejidad para la resección, por lo que se decidió continuar el tratamiento médico y realizar un nuevo control por imagen. Con la antibioterapia instaurada, la paciente presentó mejoría clínica y analítica con defervescencia de la fiebre, resolución del dolor abdominal y disminución de la PCR de 25 a 0,5 mg/dL.
En la RM de control realizada 11 días después, se objetivó disminución del tamaño de la lesión a expensas de colección líquida en su interior (la colección pasó de medir 7 × 5 × 2,6 cm a medir 5,3 × 4,4 × 1,3 cm). En la exploración abdominal la masa fue progresivamente menos perceptible a la palpación.
Atendiendo a la reducción de la lesión se desestimó la necesidad de tratamiento quirúrgico y dada la evolución clínica y analítica favorables, se procedió al alta hospitalaria tras 20 días de ingreso.
S. typhimurium es un bacilo gramnegativo anaerobio facultativo que pertenece a la subespecie de Salmonelas gastroenteríticas, cuyo mecanismo principal de transmisión es por la ingesta de agua o alimentos contaminados (huevos y carnes poco cocinadas, mayoritariamente). Tras la ingesta, las bacterias invaden el epitelio de la mucosa del intestino delgado y del colon, pudiendo así producir gastroenteritis o bien producir bacteriemia y metastatizar en cualquier parte del organismo y formar abscesos (como el hepático y el esplénico, entre otros), o bien producir endocarditis o infección de un aneurisma o de material protésico vascular. En el caso de esta paciente, la vía más probable fue a través del drenaje linfático intestinal.
Respecto al tratamiento antibiótico realizado, una vez identificado el microorganismo y teniendo en cuenta la elevada complejidad de la resección quirúrgica y el tamaño de la colección, se optimizó el esquema antibiótico a cefotaxima 2 g/4 h en combinación con ciprofloxacino 400 mg/8 h, para alcanzar concentraciones plasmáticas elevadas y una mayor difusión al interior de la colección abdominal.
Tras el cambio del tratamiento antibiótico, la paciente quedó afebril y evolucionó favorablemente, por lo que a las 2 semanas se modificó a cefixima 400 mg/24 h + ciprofloxacino 500 mg/12 h por vía oral durante 6 semanas, simplificando posteriormente a ciprofloxacino 500 mg/12 h durante 8 semanas más. S. typhimurium puede desarrollar bacterias persistentes, que pasan a un «estado de no crecimiento» y, por tanto, son menos sensibles a los tratamientos recibidos. Por este motivo, se priorizó el empleo de quinolonas y se prolongó el tratamiento hasta completar un total de 4 meses de antibioterapia dirigida.
Tras el alta hospitalaria, la paciente realizó seguimiento ambulatorio con control por imagen por parte del Servicio de Enfermedades Infecciosas. La paciente completó un total de 4 meses de tratamiento antibiótico dirigido, que se retiró al observar la resolución de las colecciones en los controles radiológicos ambulatorios.
Actualmente, la paciente continúa en seguimiento con controles clínicos y radiológicos semestrales. En el último control a los 2 años y medio del ingreso, el linfangioma mide 2,7 × 3,7 cm en el plano axial, aunque continúa contactando con los vasos mesentéricos (fig. 4). La paciente permanece asintomática a la fecha de redacción del presente caso clínico.
 para valoración comparativa de la lesión quística polilobulada que se extiende por la raíz mesentérica, en el periodo de un año. A y C) RM potenciada en T2. B y D) RM potenciada en T1 con contraste endovenoso extracelular (gadolinio). La localización mesentérica de la lesión se determina por su relación con la arteria mesentérica superior (flecha gris con borde blanco) y la vena mesentérica superior (flecha blanca). La valoración comparativa muestra una franca disminución del tamaño de la lesión (enmarcada por triángulos blancos) y la resolución de la hipercaptación de las paredes que la limitan.")
RM tras 2 años y medio de seguimiento. Se muestran 4 imágenes de RM del hemiabdomen superior, (cortes axiales) para valoración comparativa de la lesión quística polilobulada que se extiende por la raíz mesentérica, en el periodo de un año. A y C) RM potenciada en T2. B y D) RM potenciada en T1 con contraste endovenoso extracelular (gadolinio). La localización mesentérica de la lesión se determina por su relación con la arteria mesentérica superior (flecha gris con borde blanco) y la vena mesentérica superior (flecha blanca). La valoración comparativa muestra una franca disminución del tamaño de la lesión (enmarcada por triángulos blancos) y la resolución de la hipercaptación de las paredes que la limitan.
Linfangioma quístico mesentérico y retroperitoneal gigante complicado con absceso por S typhimurium.
Responsabilidades éticasSe obtuvo el consentimiento informado de la paciente para la publicación del artículo.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
