




A escala mundial, la incidencia de cáncer colorrectal en ambos sexos presenta un claro gradiente según el grado de industrialización y la latitud. Las zonas más desarrolladas y situadas hacia el norte, presentan tasas más elevadas1.
La mortalidad por cáncer colorrectal en España es menor que la de los países del norte de Europa, siendo la variabilidad interprovincial muy baja2. En el período 1997-2008 se registraron en Cantabria un total de 4.081 casos de cáncer colorrectal invasivo, con un promedio anual de 340,08 casos. El cáncer de colon, con 2.606 casos (217 casos/año), fue más frecuente que el de recto+unión colorrectal, con 1.475 (123 casos/año). Estas cifras absolutas suponen una tasa de incidencia ajustada de 30,15 casos/100.000 habitantes2.
Este cáncer se ha relacionado con genes encargados de la reparación de los errores de emparejamiento del ADN (Mis Match Repair [MMR]), que son: MSH2, MSH6, MLH1 y PMS2. La presencia de dichos genes, supone una mayor predisposición al desarrollo de cáncer colorrectal y otras neoplasias relacionadas (síndrome de Lynch), entre los que destaca el cáncer de endometrio en mujeres de edad joven3,4.
El diagnóstico definitivo se confirma mediante un estudio genético que es completo y costoso, por lo que se recomienda realizar una selección previa de pacientes5–7.
Con el fin de seleccionar a estos pacientes, se utilizan 2 criterios clínicos diferentes:
En primer lugar, los de Amsterdan II:
- a)
Tres o más individuos con cáncer colorrectal o tumor asociado al síndrome de Lynch (endometrio, estómago, intestino delgado, tracto urinario, ovario, páncreas, tracto biliar, cerebral o próstata).
- b)
Uno de ellos familiar de primer grado de los otros 2.
- c)
Afectación de 2 generaciones consecutivas, existiendo como mínimo un caso diagnosticado y confirmado antes de los 50 años1,8,9.
En segundo lugar, los criterios de Bethesda revisados:
- a)
Pacientes con cáncer colorrectal diagnosticado antes de los 50 años.
- b)
Pacientes con cáncer colorrectal sincrónico o metacrónico u otro tumor asociado al síndrome de Lynch (independientemente de la edad del diagnóstico).
- c)
Pacientes con cáncer colorrectal con histología característica de síndrome de Lynch (linfocitos infiltrantes del tumor, reacción de tipo Crohn, diferenciación mucinosa/anillo de sello o crecimiento medular), diagnosticado antes de los 60 años.
- d)
Pacientes con cáncer colorrectal y uno o más familiares de primer grado con un tumor asociado al síndrome de Lynch, uno de ellos diagnosticado antes de los 50 años.
- e)
Pacientes con cáncer colorrectal y 2 o más familiares de primer o segundo grado con un tumor asociado al síndrome de Lynch independientemente de la edad.
Los pacientes afectos deben ser seguidos con colonoscopia cada 1-2 años.
Existen 2 variantes del síndrome de Lynch. La primera, el síndrome de Turcot tipo 1 con patrón de herencia autosómica recesiva. Su principal característica son los gliomas, mientras que la poliposis es secundaria. Se ha relacionado con genes MMR encargados de la reparación de los errores de emparejamiento del ADN como: MSH2, MSH6, MLH1 y PMS2. La segunda variante, el síndrome de Muir-Torre es un cuadro raro, que se transmite por herencia autosómica dominante, cuyos genes implicados son MLH1, MSH2 y MSH6. Se asocia a neoplasias en colon, mama, aparato genitourinario y alteraciones cutáneas como tumores basocelulares, de células escamosas o tumores sebáceos8,9.
Se presenta el caso de una paciente diagnosticada de adenocarcinoma de colon derecho a los 74 años, con antecedentes de cáncer en familiares de primer grado.
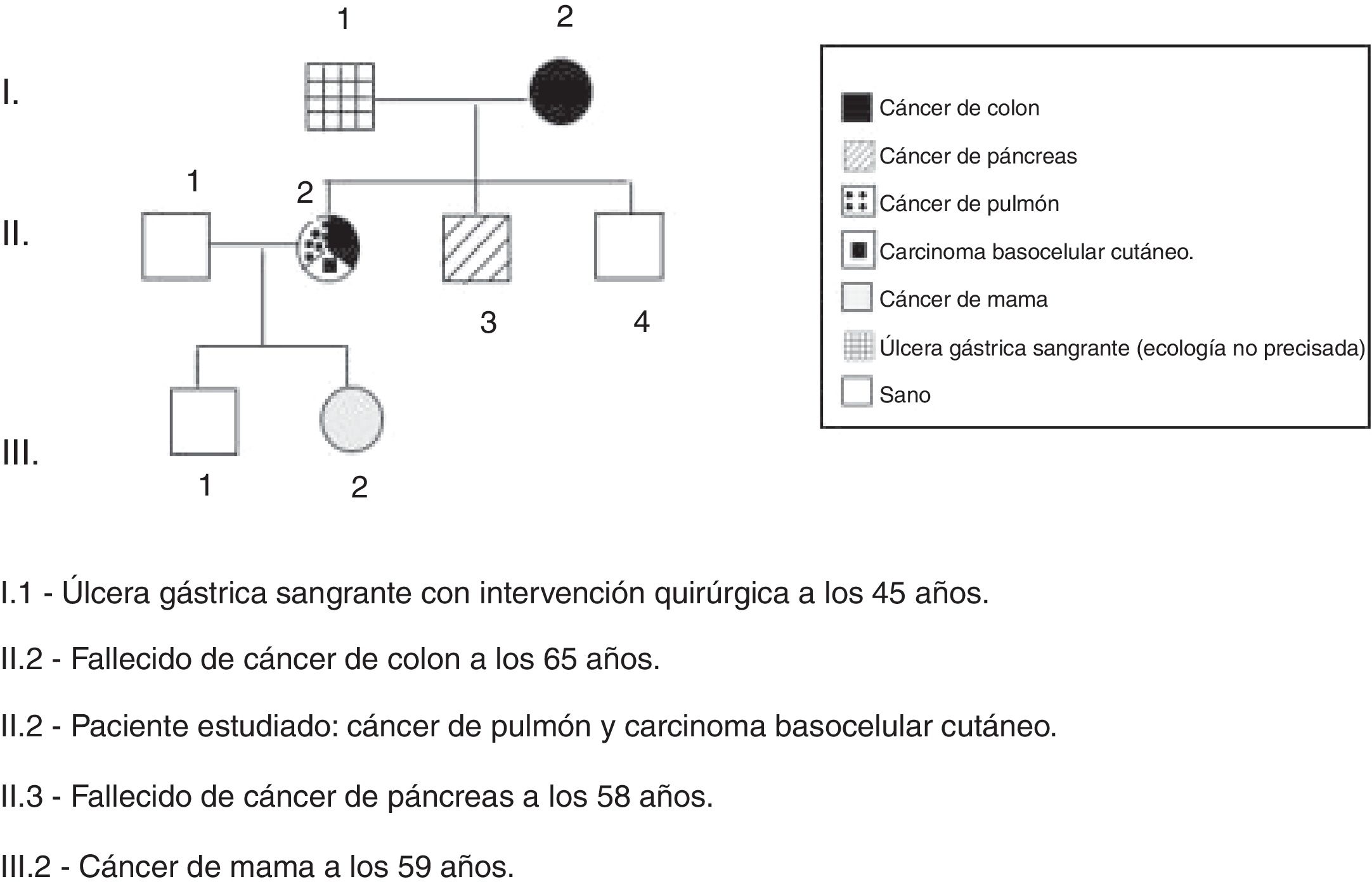
Caso clínicoPaciente mujer de 84 años de edad, con antecedentes personales de hipertensión arterial e hipotiroidismo; intervenida de carcinoma basocelular nasal (2013) y colecistectomía (1987). Como antecedentes familiares: madre fallecida por cáncer de colon a la edad de 66 años; hermano fallecido por cáncer de páncreas a los 58 años; hija con cáncer de mama a la edad de 59 años y padre con antecedentes de úlcera gástrica sangrante, intervenida quirúrgicamente a los 45 años sin etiología precisada (se adjunta diagrama genético explicativo: fig. 1).

En febrero de 2007 se realiza hemicolectomía derecha, cuya anatomía patológica fue de adenocarcinoma de colon derecho pT3pN1 (1/29) M0. El estudio genético reveló mutaciones en los genes MSH2, MSH6, MLH1 y PMS2. En marzo de 2008 fue diagnosticada por mediastinoscopia más biopsia de adenopatía 7, PET y TAC, de adenocarcinoma de pulmón T2N2M0. Recibió tratamiento con 3 ciclos de cisplatino-gemcitabina, objetivándose respuesta parcial. Posteriormente fue sometida a lobectomía inferior derecha más linfadenectomía. En seguimiento sin evidencia de enfermedad hasta agosto de 2015 cuando, mediante TAC, PET, BAG y posterior resección atípica se diagnostica de adenocarcinoma de pulmón en LSD pT1N0M0.
DiscusiónLos pacientes con síndrome de Lynch tienen un riesgo mayor de presentar tumores en diferentes órganos a lo largo de su vida2: 52-82% en el caso de cáncer de colon y recto (edad media al diagnóstico: 44-61 años); 25-60% de cáncer de endometrio (edad media al diagnóstico: 48-62 años); 6-13% de neoplasias de estómago (edad media al diagnóstico: 56 años); 4-12% de cáncer de ovario (edad media al diagnóstico: 42,5 años). El riesgo de otros tumores relacionados con el síndrome de Lynch es menor, pero aumenta cuando se compara con el existente en la población general. La herencia es autosómica dominante. La presencia de mutaciones en los genes referidos anteriormente implica un riesgo mayor de neoplasias1,7.
Existen 2 tipos de pruebas de despistaje de tumores para el síndrome de Lynch4: la inestabilidad de microsatélites (MSI) y la inmunohistoquímica (IHC). Ambas buscan cambios en los genes o proteínas que se relacionan con el síndrome de Lynch y predicen si existe un riesgo aumentado de desarrollar un tumor nuevo. La mejor forma de diagnosticar el síndrome es mediante las pruebas genéticas. Si se encuentra una mutación en un gen se realizan pruebas en los otros familiares para saber si también portan la mutación que predispone a dicho síndrome10.
En el caso estudiado, existían antecedentes en familiares de primer grado de cáncer de colon, páncreas y mama en madre, hermano e hija, respectivamente. Además, su padre fue intervenido quirúrgicamente por úlcera gástrica sangrante de etiología no precisada. La paciente presentó un adenocarcinoma de colon derecho, un carcinoma basocelular de piel y un adenocarcinoma de pulmón, con un estudio inmunohistoquímico que reveló la expresión de los genes MSH2, MSH6, MLH1 y PMS2. Aunque el cáncer de pulmón no es una localización habitual de tumores en el síndrome de Lynch, estos individuos tienen un riesgo aumentado respecto a la población general de desarrollar cáncer7.
Este caso aporta una gran reflexión a todos los médicos de atención primaria, pues manifiesta la gran oportunidad de detectar enfermedades con agregación familiar, al trabajar en el primer nivel asistencial y tener acceso al entorno del paciente.
Otra reflexión a tener siempre en mente es que determinadas enfermedades, síntomas o signos de una afección pueden constituir síndromes familiares. De esta manera se puede contribuir desde la atención primaria al diagnóstico precoz de la enfermedad potencialmente tratable o evitable. Los profesionales deben ser conscientes de que lo esencial en el ejercicio de la práctica médica habitual no es tanto poseer conocimientos absolutos de las diferentes enfermedades, como tener al menos la inquietud de una sospecha diagnóstica que lleve a indagar y a detenerse en ciertos casos para llegar a un diagnóstico certero.
Es inherente a la especialidad la concepción de salud y enfermedad de forma global y familiar y, con una luz e intuición apropiadas y aprendidas, estar alerta ante los casos familiares de enfermedad.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Los autores agradecen al Dr. José Luis López por su oportuna crítica, y a la paciente por su cooperación y predisposición.
