




INTRODUCCIÓN
El ácido acetilsalicílico (AAS) es el tratamiento antiagregante más empleado en la práctica clínica. Además, es el fármaco más coste-efectivo en la prevención secundaria de enfermedad cardiovascular, pero los fallos en el tratamiento son relativamente frecuentes1.
Desde hace más de una década se sabe que el tratamiento antiagregante con AAS reduce el riesgo de muerte de origen cardiovascular en torno a un 15-20% y el riesgo de infarto de miocardio (IAM) y de accidente cerebrovascular (ACV) no fatales en alrededor de un 30-35% de pacientes con angina inestable, sospecha de IAM agudo o antecedente de IAM, ACV o episodio isquémico cerebral transitorio2.
Su efectividad ha sido bien establecida en ensayos clínicos y metaanálisis con una reducción aproximadamente del 25% de las recurrencias1.
No obstante, los efectos antitrombóticos del AAS son muy variables, ya que se producen entre un 5 y un 60% de fracasos terapéuticos a la hora de alcanzar un nivel adecuado de inhibición plaquetaria3 y entre el 10 y el 20% de los pacientes tratados puede desarrollar recurrencias de eventos vasculares4.
Se define como resistencia al AAS la recurrencia de eventos vasculares en estos pacientes5-7. De forma confusa este término ha sido usado en la literatura para describir un fenómeno tanto bioquímico como clínico. La resistencia al AAS hace referencia a la persistencia de la activación plaquetaria demostrada mediante prueba de función plaquetaria (resistencia al AAS bioquímico) o a la recurrencia de eventos cardiovasculares (resistencia al AAS clínico). El concepto clínico no es específico y puede ser preferible etiquetarlo como fallo clínico al tratamiento8. Sanderson et al definen la resistencia al AAS como el fenómeno bioquímico que puede tener consecuencias clínicas1.
MECANISMO DE ACCIÓN DEL ÁCIDO ACETILSALICÍLICO
Agregación plaquetaria
El AAS produce una inhibición selectiva de la ciclooxigenasa (COX) que ayuda a catalizar la síntesis de prostaglandinas y desempeña un importante papel en la pared del vaso. Hay 2 tipos de COX: 1 (COX-1) y 2 (COX-2).
La COX-1 se expresa regularmente en las plaquetas maduras y cataliza la síntesis de tromboxano A2 (Tx-A2), que favorece la agregación plaquetaria y la vasoconstricción.
El Tx-A2 es sintetizado y liberado por las plaquetas en respuesta a numerosos estímulos, por ejemplo colágeno, epinefrina, adenosín difosfato y trombina. Además, sus niveles aumentan en situaciones de agregación plaquetaria tales como ángor inestable, IAM y ACV.
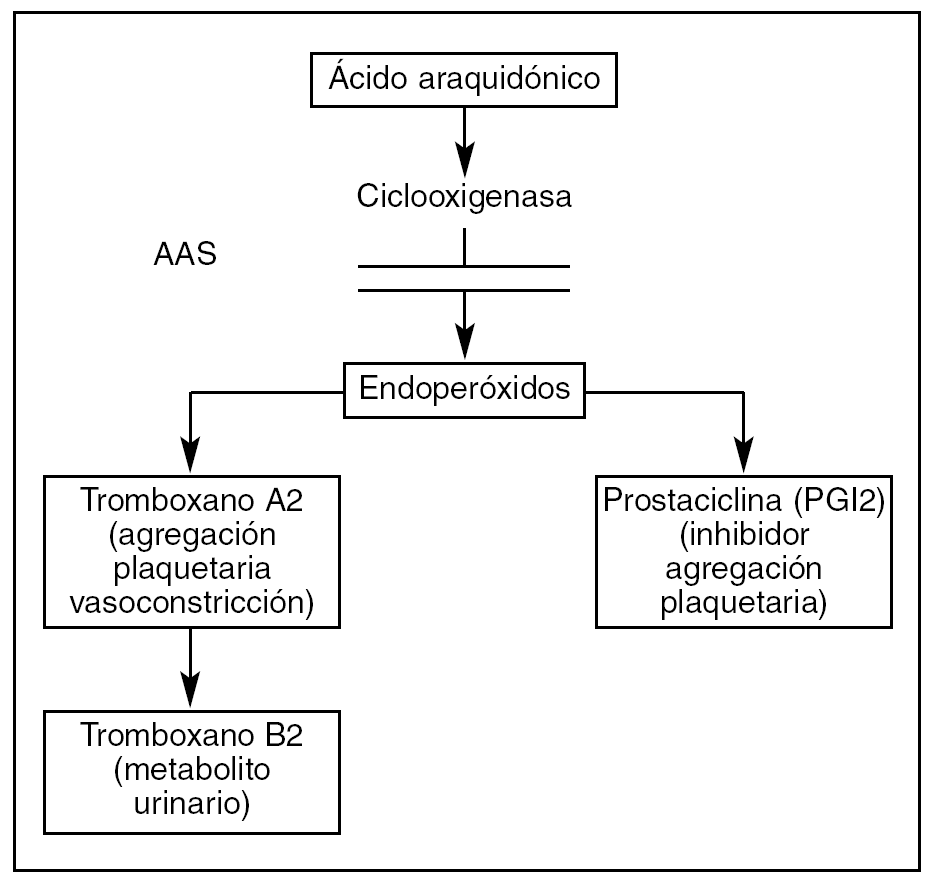
Por otro lado, la COX-2 cataliza la síntesis de prostaciclina (PGI2), un potente inhibidor de la agregación plaquetaria producida en el endotelio celular (fig. 1).
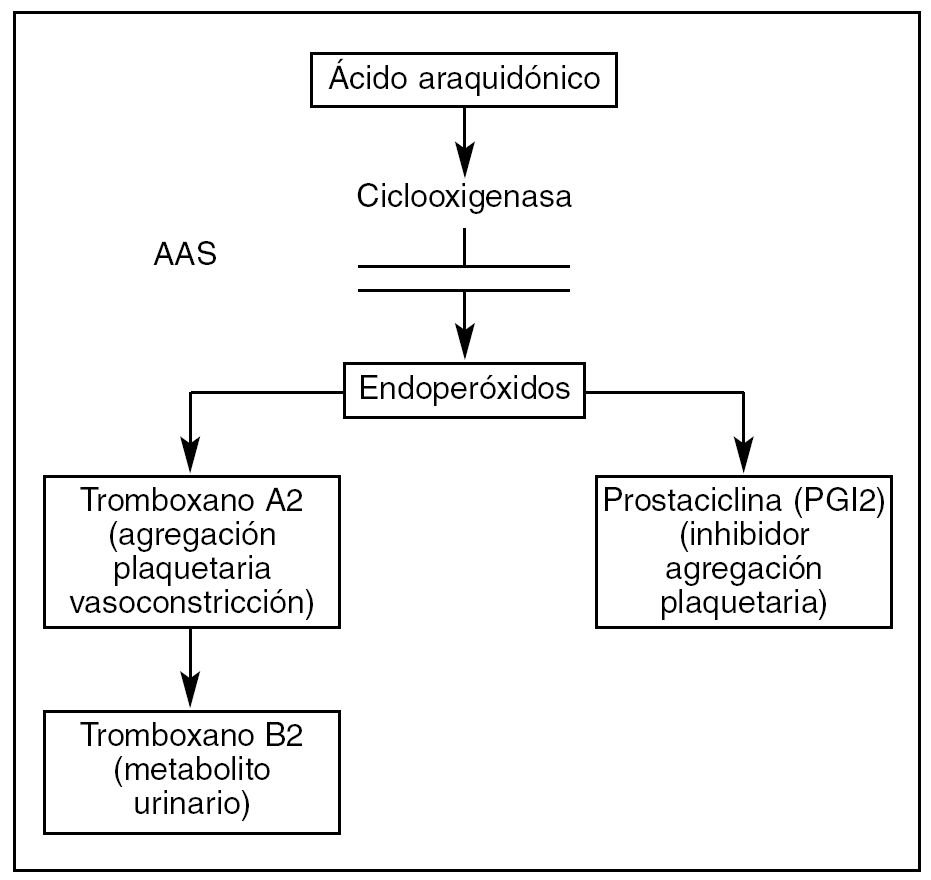
Figura 1. Papel del ácido acetilsalicílico (AAS) en la agregación plaquetaria.
La COX-2 rara vez se encuentra en las plaquetas maduras pero se ha encontrado en las jóvenes durante los periodos de recambio plaquetario rápido.
A diferencia de otros antiinflamatorios no esteroideos (AINE), el AAS inhibe la COX-1 aproximadamente 170 veces más que la 2; por ello el AAS inhibe el Tx-A2 mucho más que la prostaciclina. El resultado es una inhibición de la activación y la agregación plaquetaria.
Mientras que la COX-1 se puede inhibir con dosis de 30 mg de AAS, para la inactivación de la COX-2 (efecto antiinflamatorio) se necesitan dosis superiores9.
Acciones del ácido acetilsalicílico
El AAS se absorbe rápidamente en el tracto gastrointestinal, alcanzando el pico plasmático a los 30-40 minutos. Aunque la vida media plasmática es de 20 minutos, puede ser tomada una vez al día, ya que se une irreversiblemente a la COX-1.
El AAS logra la inhibición de la COX-1 y ésta permanece inactiva durante el resto de la vida de la plaqueta. De este modo reduce la síntesis de prostaglandinas durante la vida media de las plaquetas (de 8 a 10 días) y, especialmente, la producción de Tx-A21.
Se han propuesto otros mecanismos adicionales del AAS sobre sus efectos clínicos en las enfermedades cardiovasculares, incluyendo la inhibición de la activación plaquetaria mediada por neutrófilos, mecanismos sobre el endotelio y efectos antioxidantes.
FRACASA EL ÁCIDO ACETILSALICÍLICO: ¿FALLO CLÍNICO O BIOQUÍMICO?
La recurrencia de eventos cardiovasculares no es infrecuente en pacientes a los que se les ha prescrito AAS para prevención secundaria.
Hay varios factores implicados en la recurrencia de dichos eventos como son: el tabaco, las interacciones medicamentosas, la falta de adherencia al tratamiento, las patologías asociadas y la resistencia al AAS.
Este término se emplea no sólo para describir la ausencia del efecto farmacológico esperado sobre las plaquetas, sino también por la no consecución de los resultados clínicos esperados, tales como la recurrencia en los eventos cardiovasculares. La resistencia al AAS quizá se pueda entender mejor como un fenómeno medible, que ocurre en pacientes en tratamiento con AAS, en los que persiste la activación plaquetaria y que puede desarrollar fallos en el tratamiento.
¿Qué factores pueden contribuir a la recurrencia?
La inadecuada adherencia al tratamiento
Hay pacientes que no toman la medicación prescrita. Tarjan et al10, en un estudio con 75 pacientes enfermos coronarios en tratamiento con AAS, encontraron que el 11% no presentaba adherencia al tratamiento.
Dosis administrada
Diversos ensayos clínicos sugieren que dosis diarias de AAS de 75 a 150 mg son óptimas para obtener un mejor equilibrio riesgo-beneficio. Otros ensayos clínicos indican que la reducción de eventos cardiovasculares con altas dosis de AAS (de 500 a 1.500 mg/día) pueden no ser mejores que bajas dosis de AAS (de 75 a 150 mg/día)2,3,11. Los efectos adversos inducidos por el AAS son dosis dependientes4.
Helgason et al12 realizaron un estudio de agregación plaquetaria en pacientes con ACV previo y concluyeron que:
1. Un grupo de pacientes que, inicialmente, presentaba una inhibición completa de la agregación plaquetaria perdía parte del efecto antiagregante a lo largo del tiempo, sin cambiar la dosis.
2. Un grupo de pacientes con inhibición parcial conseguía una inhibición completa aumentando la dosis aunque, parte de ellos, posteriormente, la perdía pasado el tiempo.
3. Hay pacientes con resistencia al AAS a pesar de dosis de 1.300 mg diarios.
Comorbilidad asociada
Los pocos estudios que han comparado el efecto en la agregación plaquetaria del AAS en fumadores y no fumadores han encontrado que fumar reduce el efecto antiagregante del AAS13,14.
Fumar, la angina inestable, la hiperlipidemia y la diabetes pueden interferir en el efecto del AAS sobre la activación plaquetaria por un incremento de la producción de isoprostanos (prostaglandina F2)13,15,16, que inducen vasoconstricción y tienen efectos protrombóticos, aumentan la respuesta de las plaquetas a otros agonistas y además "insensibilizan" la acción del AAS sobre la biosíntesis de tromboxano8,17.
Estudios en fumadores han demostrado que la producción de isoprostanos aumenta con el número de cigarrillos fumados y que 75 mg al día de AAS pueden no tener el efecto deseado en estos pacientes14,18. Este mecanismo podría justificar la recurrencia de eventos vasculares a pesar del empleo de dosis preventivas de AAS.
Otras condiciones, como arteritis o embolismos cardiacos procedentes de prótesis valvulares, afectación cardiaca reumática o endocarditis infecciosa, pueden ser causa independiente de recurrencias en pacientes tratados con AAS1.
Interacciones medicamentosas
El consumo regular de ciertos AINE como el ibuprofeno y la indometacina parece antagonizar el efecto antiplaquetario del AAS19-21.
El ibuprofeno y el naproxeno pueden interferir con dosis bajas de AAS, ya que evitan la acetilación irreversible de la COX-1; este fenómeno no se observa con los inhibidores selectivos de la ciclooxigenasa (coxib) o el diclofenaco, que tiene mayor selectividad por la COX-222.
¿Qué es la resistencia al AAS?
La resistencia a los fármacos es el resultado de los cambios producidos en virus, microbios o células cancerígenas de forma que reducen o eliminan la efectividad de los agentes empleados para curar o prevenir infecciones o enfermedades23. También pueden producirse cambios genéticos que afecten a los receptores del fármaco. La exposición al medicamento es generalmente el desencadenante.
La llamada "resistencia" al AAS difiere de la definición clásica de resistencia a un fármaco ya que el cambio no se produce en el receptor, los efectos pueden variar a lo largo del tiempo y son parcialmente reversibles incrementando la dosis12.
Aunque las plaquetas jóvenes expresan tanto COX-1 como COX-2, las plaquetas maduras expresan sólo COX-1. Mientras la COX-1 es altamente sensible a bajas dosis de AAS, la COX-2 se inhibe sólo con dosis lo suficientemente altas como para proporcionar efectos analgésicos o antiinflamatorios24.
La administración crónica de AAS aumenta la proporción de plaquetas jóvenes, lo que podría explicar los cambios a lo largo del tiempo de la sensibilidad plaquetaria a la inhibición a dosis bajas del fármaco24.
Un pequeño porcentaje de resistencias al AAS podría explicarse por la existencia de un polimorfismo en la expresión de la COX-1 que afecta al receptor, tal como se describe en la definición clásica de resistencia farmacológica, sin embargo, no explicaría los cambios a lo largo del tiempo.
Aunque se ha descrito esta variabilidad genética, tanto el aumento como la disminución de la respuesta al AAS se asocian al mismo haplotipo de COX-1, presente en el 12% de la población25,26.
PRUEBA DE FUNCIÓN PLAQUETARIA
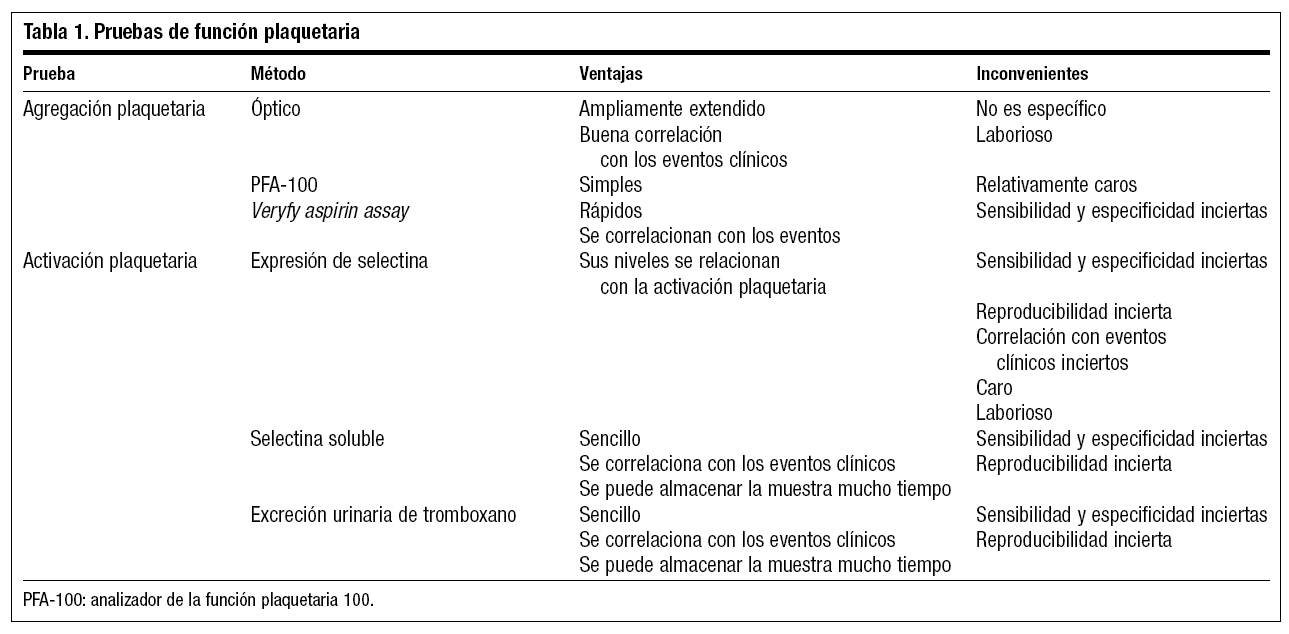
Aunque existen muchas pruebas para valorar la función plaquetaria, se pueden agrupar en aquellas que miden la agregación y las que miden la activación plaquetaria. En general las primeras cuantifican la agregación en respuesta a agonistas conocidos, mientras que las pruebas de activación miden sustancias en plasma y orina que se liberan al activar las plaquetas (tabla 1).
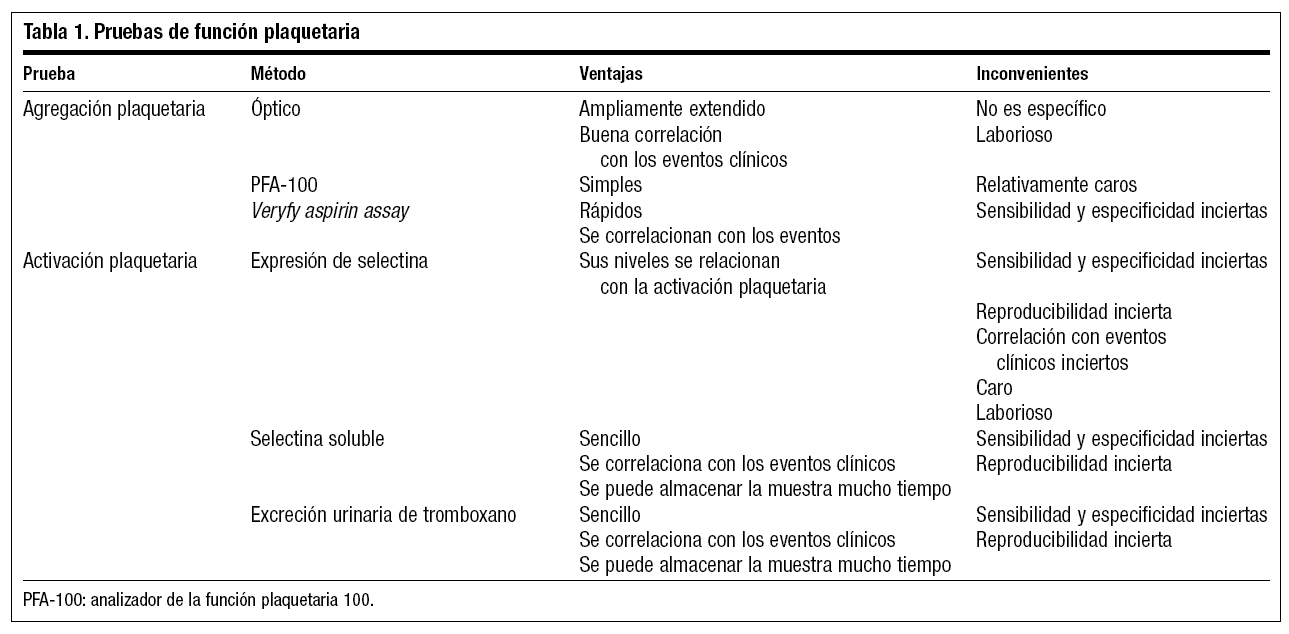
Pruebas de agregación plaquetaria
Agregómetro de función plaquetaria en plasma (método óptico)
Es una de las pruebas más antiguas; cuantifica la agregación plaquetaria basada en la cantidad de luz transmitida a través de la muestra. Esta prueba es muy laboriosa, dado que requiere un rápido procesado de la muestra en las horas siguientes a la extracción.
Medición de la agregación plaquetaria mediante impedancia eléctrica
En esta técnica no se necesita preparar el plasma. Su resultado no se correlaciona con el anterior.
Analizador de la función plaquetaria 100 (PFA-100) o Veryfy now aspirin assay
Simula in vitro la hemostasia primaria en sangre periférica. La mayoría de los estudios revisados están de acuerdo en que el analizador de la función plaquetaria 100 (PFA-100) es un método simple, real y reproducible para medir la función plaquetaria in vitro27-34.
Pruebas de activación plaquetaria
La ventaja frente a las pruebas anteriores es que pueden usarse en muestras almacenadas.
Tromboxano-B2 en orina
Es el producto final del ciclo del ácido araquidónico y refleja el nivel de la actividad plaquetaria35,36. Esta prueba es relativamente sencilla y barata y ha sido utilizada en diversos estudios sobre resistencia al AAS. La administración de AAS reduce su excreción urinaria de un 60 a un 80%. Sin embargo, podemos encontrar este metabolito en orina tras la administración de AAS debido a que las plaquetas no son su única fuente. No obstante, es un método fiable.
Eikelboom et al37 apreciaron que el riesgo de IAM, ACV o muerte cardiovascular aumenta a mayor nivel detectado de tromboxano B2 (Tx-B2) en orina. Una limitación a este estudio fue la dificultad para diferenciar si los resultados eran debidos a la falta de cumplimiento terapéutico, al empleo de AINE o a la insensibilidad al AAS.
Tromboxano B2 en suero
La administración de 100 mg de AAS en individuos sanos reduce un 98% la actividad plaquetaria de la COX-1, y a su vez los niveles de Tx-B238.
Expresión de P-selectina en la membrana plaquetaria
Aumenta con la activación y degranulación plaquetaria. Su medición es cara y compleja39.
P-selectina soluble en plasma
Su elevación indica un aumento de la activación plaquetaria40. Este método es más sencillo pero relativamente caro.
La utilización de estas pruebas podría emplearse en la práctica diaria para ayudar en nuestra toma de decisiones, pero la relevancia de las diversas pruebas plaquetarias es desconocida. Además, la correlación entre los resultados de las diferentes pruebas de medición son pobres, por lo que no se recomienda emplear ninguna prueba de función plaquetaria para valorar qué individuos van a responder al AAS. La Seventh American College of Chest Physicians Consensus Conference on Antithrombotic and Thrombolytic Therapy, la European Society Of Cardiology Task Force on Antiplatelets y el Platelet Physiology Subcommittee of The Scientific and Standardization Committee of the International Society on Thrombosis and Haemostasis indican unánimemente que, en pacientes individuales, ninguna prueba de función plaquetaria es adecuada para valorar el efecto del AAS24.
CONCLUSIONES
La resistencia al AAS parece ser el resultado de una inhibición incompleta de la función plaquetaria, situación que puede fluctuar a lo largo del tiempo.
Las pruebas para medir la resistencia al AAS y los cambios terapéuticos basados en estas pruebas de laboratorio no se recomiendan debido a la falta de datos sobre la eficacia y la seguridad de esta estrategia.
Desde nuestra consulta, tenemos que hacer hincapié en los factores potencialmente limitantes del efecto antiplaquetario del AAS, como son la falta de adherencia al tratamiento y las interacciones farmacológicas, especialmente con ibuprofeno, naproxeno e indometacina. Estas medidas pueden mejorar la eficacia clínica.
Son necesarios más estudios para establecer cuáles son los métodos que mejor identifican a aquellos pacientes con alto riesgo de desarrollar eventos cardiovasculares a pesar de estar en tratamiento con AAS y estudios para determinar la influencia de la dosis en estas resistencias.
Correspondencia:
Correo electrónico: drodriguez.gapm04@salud.madrid.org
Recibido el 20-11-07; aceptado para su publicación el 18-12-07.
