





Tradicionalmente, los términos polimiositis (PM) y dermatomiositis (DM) se utilizan para definir una serie de enfermedades sistémicas que se caracterizan por miopatía inflamatoria mediada por mecanismos autoinmunitarios1,2. En realidad, estas dos enfermedades forman parte del amplio grupo de las miopatías inflamatorias idiopáticas. Estas enfermedades inflamatorias musculares son muy infrecuentes y los estudios epidemiológicos indican que presentan una incidencia anual de 1-10 casos por millón de habitantes y un ligero predominio de la afección en mujeres (2,5:1); aunque pueden aparecer a cualquier edad, son más frecuentes en la infancia y entre la cuarta y la quinta décadas de la vida1,2.
Su diagnóstico se realiza habitualmente aplicando los criterios descritos por primera vez por Bohan et al3 (tabla 1): debilidad muscular progresiva, elevación de las enzimas musculares, patrón electromiográfico miopático, evidencia de infiltrado inflamatorio en la biopsia muscular y lesiones cutáneas características de DM. Se considera que el diagnóstico de PM es definitivo si los primeros cuatro criterios están presentes, probable si hay tres y posible si hay dos. Para el diagnóstico de DM siempre debe haber lesiones dérmicas características y al menos uno de los primeros cuatro criterios.

Tabla 1. Criterios diagnósticos de polimiositis de Bohan et al3
En el 20% de los adultos en que se desarrolla PM o DM del adulto, la enfermedad tiene relación con un tumor maligno, que habitualmente no está diagnosticado1. Los tumores que se asocian con el desarrollo de estas enfermedades son los habituales de los adultos; entre los más frecuentes, en algunas series, son el carcinoma de ovario en las mujeres y el de pulmón en los varones1,2.
La PM y la DM son enfermedades multisistémicas que con frecuencia afectan a otros órganos1,2,4. La afección pulmonar es de las más frecuentes y en algunas series hasta el 61% de los enfermos la tienen y es una de las principales causas de morbilidad y mortalidad en esta enfermedad4-7. Habitualmente es una neumonitis intersticial que sin tratamiento evoluciona a la fibrosis pulmonar y ensombrece el pronóstico de los enfermos. Otras manifestaciones pulmonares son la insuficiencia respiratoria y las neumonías aspirativas por la debilidad muscular, la hipertensión pulmonar, la bronquiolitis obliterante, la hemorragia difusa pulmonar y los procesos infecciosos secundarios a la inmunodeficiencia originada por los fármacos empleados en el tratamiento de la enfermedad6.
Habitualmente la PM se manifiesta por la típica debilidad muscular proximal simétrica y progresiva, acompañada con frecuencia de artralgias, Raynaud y síntomas constitucionales. Aunque la afección pulmonar puede aparecer en cualquier momento de la evolución de la enfermedad, es infrecuente que ésta sea la primera manifestación. Recientemente hemos atendido a un paciente que presentó como manifestación inicial una neumonitis intersticial, que por sus inhabituales características creemos oportunos comunicar.
CASO CLÍNICOVarón de 62 años, bebedor de 60-70 g de etanol diarios y fumador de 40 cigarrillos/día, con criterios de enfermedad pulmonar obstructiva crónica (EPOC). Acudió a consulta de Atención Primaria porque presentaba desde 3 días antes cuadro de tos, expectoración blanca y fiebre de 38 °C. A la exploración física presentaba buen estado general y en la auscultación pulmonar roncus y sibilancias bilaterales. Diagnosticado inicialmente de infección respiratoria, se instauró tratamiento con amoxicilina y paracetamol orales y salmeterol inhalado. Acudió de nuevo 5 días después por empeoramiento del cuadro y presentaba disnea de esfuerzo y expectoración levemente hemoptoica. Se realizó un estudio analítico que mostró velocidad de sedimentación globular (VSG) de 28 mm/h, hemoglobina (Hb) 9,7 g/dl y 17.800 leucocitos/mm3 (el 82% neutrófilos). La gasometría arterial mostró presión de O2 en 56 mmHg, presión de CO2 en 34 mmHg y saturación de O2 del 92%; los parámetros bioquímicos del autoanalizador fueron normales, incluido el perfil hepático; no se determinó la concentración de creatinquinasa (CK). El estudio radiográfico de tórax mostró una imagen nodular compatible con absceso pulmonar a nivel de segmento 6 derecho y aumento difuso de la trama intersticial de predominio basal.
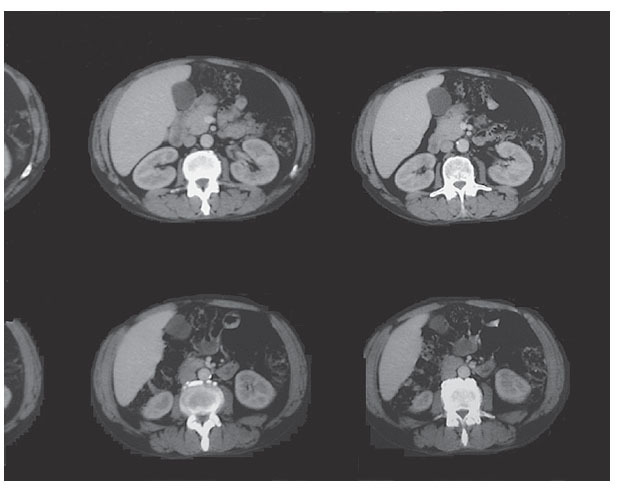
Se remitió al paciente al servicio de Urgencias hospitalario, desde donde ingresó en el servicio de Neumología con el diagnóstico de posible absceso pulmonar e insuficiencia respiratoria parcial; se inició tratamiento intravenoso con amoxicilina-clavulánico, gentamicina, broncodilatadores inhalados y esteroides a dosis de 60 mg diarios, con buena evolución clínica. Durante el ingreso se realizó tomografía computarizada (TC) de tórax, con el siguiente resultado: en el lóbulo inferior derecho se apreciaba área de infiltrado pulmonar con dos cavidades que presentaban nivel hidroaéreo en su interior, que podrían corresponder a proceso neoplásico y/o absceso pulmonar. En el medias-tino se apreciaban algunas adenopatías que alcanzaban algún centímetro de diámetro, en especial paratraqueales derechas, precarinales y subcarinales, que podían corresponder a cambios inflamatorios. Enfisema bulloso de tipo paraseptal que predominaba en los lóbulos superiores (fig. 1).
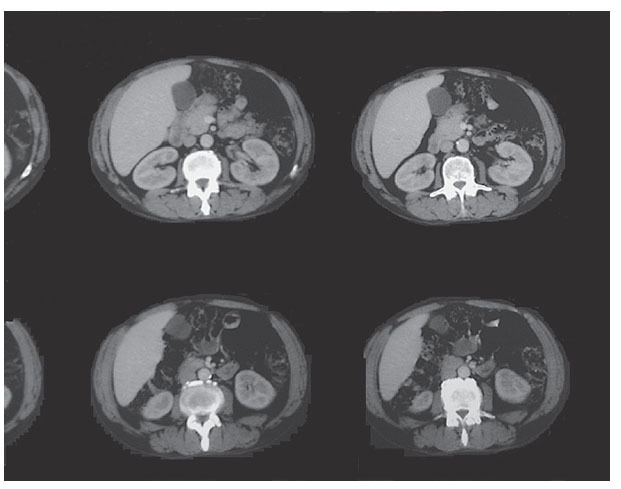
Figura 1. Tomografía computarizada: infiltrado pulmonar con cavidades en su interior.
En los días posteriores se realizó una fibrobroncoscopia, con abundantes secreciones purulentas; no se observaban lesiones endobronquiales. En la citología del cepillado bronquial, se observaron atipias sospechosas de carcinoma no microcítico, y en la biopsia transbronquial, se observó inflamación crónica inespecífica. Posteriormente se realizó nueva fibrobroncoscopia, en la que se observaron de nuevo atipias citológicas sospechosas de lesión tumoral con diferenciación escamosa. El paciente fue diagnosticado de neumonía necrosante versus absceso pulmonar y alta sospecha de carcinoma no microcítico centrado en lóbulo inferior derecho, por lo que fue derivado al servicio de Cirugía Torácica, donde luego le practicaron una lobectomía inferior derecha. El resultado histológico del lóbulo extirpado fue de pared quística fibrosa, en cuya luz se identificaban restos de tejido pulmonar y en cuya periferia el parénquima pulmonar mostraba imágenes de neumonía en resolución con tapones fibroblásticos, inflamación linfocitaria, acumulaciones de histiocitos intraalveolares y focos de hemorragia reciente; no se observaban signos de transformación tumoral en ningún nivel y había infiltrados inflamatorios linfocitarios en la periferia. En los ganglios linfáticos extirpados se observó histiocitosis sinusal, antracosis y granulomas epitelioides.
Tres meses después el paciente refirió haber mejorado notablemente de su disnea, pero persistía importante astenia y tenía mialgias, poliartralgias y debilidad muscular proximal en las cuatro extremidades, sin fiebre, pérdida de peso ni anorexia. Se realizó un control analítico en el que destacaron: VSG 89 mm/h; Hb 10,3 g/dl; volumen corpuscular medio 89 fl; ferritina 667 ng/ml; hierro 31 pg/dl; transferrina 106 mg/dl; anticuerpos antinucleares > 1/160 (patológico > 1/160); anticuerpos contra el antígeno SS-A positivos, y anticuerpos contra el antígeno Jo-1 positivos. Concentraciones séricas de CK 439 U/l; fracción MB de la CK 21 U/l, y lactatodeshidrogenasa 494 U/l.
Se derivó al paciente al servicio de Reumatología donde, tras solicitar un electromiograma, que mostró afección miopática de los músculos explorados –deltoides derecho y cuádriceps femoral derecho–, fue diagnosticado de polimiositis con afección pulmonar en el contexto. Se inició tratamiento con metotrexato (15 mg/semana) y prednisona (60 mg/día), con lo que se obtuvo mejoría de la debilidad y de los síntomas constitucionales y se normalizaron los reactantes de fase aguda y las enzimas musculares. Tres meses después de iniciado el tratamiento, el enfermo se reincorporó a su trabajo habitual de albañil y 8 meses más tarde continuaba asintomático en tratamiento con 20 mg/ semanales de metotrexato y 7,5 mg/diarios de prednisona, sin que aparecieran indicios de un tumor maligno relacionado y habiendo desaparecido los infiltrados intersticiales en la TC de control.
DISCUSIÓNLa afección pulmonar en la PM y la DM se produce en un 5-61% de los pacientes2,4,7 y puede aparecer antes, durante o después del diagnóstico de la afección muscular. Habitualmente aparece cuando la enfermedad muscular está ya diagnosticada y se trata de una neumonitis intersticial de predominio inicial en bases, que sin tratamiento evoluciona en meses hacia la fibrosis pulmonar, con severa y progresiva insuficiencia respiratoria e incluso muerte. La extensión y la severidad de la miositis no se correlacionan con la aparición de enfermedad pulmonar5. Los métodos de elección para diagnóstico de la afección pulmonar son TC de alta resolución y pruebas funcionales respiratorias (PFR). Está especialmente recomendado que estas pruebas formen parte de la evaluación inicial de los pacientes con PM/DM y durante su seguimiento5. En alrededor del 30% de los pacientes con PM se desarrollan anti-Jo-1. Estos anticuerpos presentan una fuerte correlación con enfermedad intersticial pulmonar en curso5, activa en casi el 100% de los pacientes con anticuerpos positivos (como es nuestro caso) y sólo en el 22% de los negativos8. Los pacientes con afección pulmonar tienen mayor mortalidad, que varía según los diferentes estudios entre el 40 y el 62%5,8, que los pacientes con PM/DM aislada. Clínicamente, se manifiesta al inicio por disnea de esfuerzo. En la radiografía puede observarse un incremento de la trama intersticial, pero es la TC lo que nos aporta más información al observarse inicialmente patrón intersticial en aspecto de vidrio deslustrado y de predominio basal, y en las fases más evolucionadas hallazgos de fibrosis pulmonar. Las PFR muestran inicialmente una disminución de la difusión pulmonar y en los casos avanzados restricción pulmonar con diferentes grados de hipoxemia. Estas pruebas sirven para seguir la evolución de la enfermedad pulmonar y la respuesta al tratamiento.
Se considera que la afección pulmonar es una grave manifestación sistémica y obliga al tratamiento con glucocorticoides e inmunosupresores2,4,9. Habitualmente se inicia el tratamiento con metotrexato a dosis de 20-25 mg/semanales y en los casos rebeldes se sustituye por ciclofosfamida oral2,4.
El caso clínico presentado corresponde a un varón de mediana edad, con antecedentes personales de EPOC, que desarrolla inicialmente una neumopatía intersticial acompañada de una neumonía abscesificada y con alteraciones citológicas que indican un posible tumor pulmonar. La afección pulmonar es la causa de que el paciente solicitara atención médica, y aunque el paciente presentaba en los últimos meses artralgias y astenia, venía realizando una vida normal, incluida su actividad laboral de albañil. Es tras la resección del lóbulo abscesificado cuando, ante la persistencia de los reactantes de fase aguda y la aparición de debilidad muscular, se plantea que alguna enfermedad del tejido conectivo sea la causa del proceso pulmonar y sistémico. Parece relevante reseñar dos aspectos de este caso; primero, ante lo que debemos tener especial precaución, que el diagnóstico de presunción en cuanto a la afectación pulmonar (previo al resultado definitivo de la anatomía patológica de la pieza quirúrgica) fue un tumor maligno pulmonar (presentaba células con atipias en la citología de dos fibrobroncoscopias sucesivas), con la consecuente decisión terapéutica que conllevó. Y segundo, que la afección pulmonar fue la manifestación inicial de la PM, una neumonitis intersticial que por sus inhabituales características (y forma de presentación) hemos considerado oportuno comunicar. Aunque lo más habitual es que las manifestaciones musculares precedan o sean simultáneas a las pulmonares, en ocasiones es la afección pulmonar la primera manifestación de la enfermedad; esto ocurre en algunas series hasta en el 30% de los casos10. No debemos olvidar que en ocasiones las enfermedades del tejido conectivo se inician con afección de diferentes órganos sistémicos antes de que las manifestaciones osteomusculares características aparezcan.
Correspondencia: L. de Prado Prieto. Centro de Salud Colmenar de Oreja. Alegas, s/n. Colmenar de Oreja. 28380 Madrid. España. Correo electrónico: lidiadeprado@hotmail.com
Recibido el 21-01-2008; aceptado para su publicación el 23-04-2008.
