







INTRODUCCIÓN
Las gammapatías monoclonales (GM) constituyen un grupo heterogéneo de trastornos que se caracterizan por la proliferación estable o progresiva de células plasmáticas, produciendo inmunoglobulinas (Ig) iguales entre sí que reciben el nombre de Ig monoclonales o componente M1,2.
La presencia de un componente proteico monoclonal en el suero constituye un hallazgo relativamente frecuente en personas de edad avanzada3. En mayores de 70 años, la prevalencia de esta alteración alcanza un 3% cuando se emplea un método convencional de electroforesis, mientras que con técnicas de electroforesis de alta resolución sobre gel de agarosa la prevalencia llega a ser del 10% en mayores de 60 años4.
El interés clínico de la detección de una proteína anómala en un proteinograma solicitado, generalmente en el estudio de un trastorno no relacionado, en la investigación de una velocidad elevada o en una revisión general, viene determinado tanto por el hecho de que dicha alteración puede ser indicativa de procesos patológicos tan dispares como una infección o una neoplasia, como por la circunstancia de que a nivel evolutivo pueda presentar comportamientos radicalmente distintos, desapareciendo en algunos casos, permaneciendo estable en otros o, por último, presentando una evolución progresiva hacia una patología maligna5.
El papel del médico de Atención Primaria se centra en la derivación del paciente que presenta una GM al hematólogo para la realización de las pruebas complementarias necesarias para realizar el correcto diagnóstico diferencial entre las distintas gammapatías, y sobre todo, una vez realizado el diagnóstico, en la colaboración con el nivel especializado en el seguimiento de estos pacientes.
EXPOSICIÓN DEL CASO CLÍNICO
Se trata de una mujer de 64 años que acude a nuestra consulta del Centro de Salud de forma programada para la revisión anual de su hipertensión arterial (HTA). La paciente se encuentra asintomática, habiendo mantenido buen control de sus cifras de presión arterial (PA) durante todo el año.
Entre los antecedentes personales de la paciente destacan: HTA en tratamiento farmacológico, poliartrosis y es portadora de anticuerpos de hepatitis B. No fuma ni bebe alcohol. Se encuentra en tratamiento con: enalapril 20 mg/día; ranitidina 150 mg/día y lorazepam 1 mg/24 horas.
Exploración física
Peso: 76 kg; talla: 1,62 m; índice de masa corporal (IMC): 29 kg/m2. PA (media de 3 determinaciones): 138/76 mmHg.
La paciente está consciente y orientada, bien hidratada y prefundida, normocoloreada y eupneica. La auscultación cardiopulmonar es normal. La exploración abdominal es normal, sin apreciarse masas ni visceromegalias. No se palpan adenopatías en ninguna localización. Los miembros inferiores no presentan edemas y los pulsos distales están presentes y son simétricos.
Pruebas complementarias
Electrocardiograma: ritmo sinusal a 65 l/min. Sin criterios de sobrecarga o hipertrofia ventricular izquierda ni alteraciones de la repolarización. Sokolow-Lyons: 22 mm.
Hemograma: hemoglobina 11,6 g/dl; hematocrito: 34,8%; volumen corpuscular medio (VCM) 91,4 fl; hemoglobina corpuscular media (HCM) 31,4 pg; plaquetas 388 x 109/l; leucocitos 7,3 x 103/microl con fórmula normal; velocidad de sedimentación globular (VSG) 59 mm/h.
Bioquímica: glucosa 99 mg/dl; creatinina 0,8 mg/dl; colesterol total: 186 mg/dl; sodio sérico: 140 mEq/l; potasio sérico: 4,87 mEq/l; calcio: 9,4 mg/dl, estando el resto de los parámetros bioquímicos dentro de la normalidad.
Analítica en orina: sedimento sin alteraciones; cociente albúmina/creatinina (media de 2 determinaciones): 15 mg/g.
En la analítica de la paciente destaca la presencia de una leve anemia normocítica y normocrómica (hemoglobina en analítica previa: 12,6 g/dl) que se acompaña de aumento de la velocidad de sedimentación. Ante este hallazgo se reinterrogó a la paciente sobre la presencia de sangrados, alteraciones del ritmo intestinal, presencia de síndrome constitucional u otros síntomas que pudieran orientarnos hacia el origen de la anemia, pero la paciente manifestó encontrarse completamente asintomática; por lo que se inició estudio de la anemia en el que solicitó estudio del hierro, ácido fólico, vitamina B12; hormonas tiroideas, proteína C reactiva (PCR), sangre oculta en heces y proteinograma. En los resultados del estudio destacaba una PCR elevada (5,3 mg/dl) y la presencia de un aumento del componente gamma en el proteinograma (proteínas totales 7,4 g/dl; albúmina 5,8 g/dl; alfa-1 3,5 g/dl; alfa-2 1 g/dl; beta 0,9 g/dl; gamma 1,7 g/dl) que una inmunoelectroforesis posterior confirmó como paraproteína IgG kappa (1.040 mg/dl).
Así, nos encontrábamos con una paciente completamente asintomática, pero que presentaba una anemia normocrómica y normocítica con aumento de los reactantes de fase aguda (VSG y PCR) y la presencia de una paraproteína IgG kappa en suero. Ante la posibilidad de encontrarnos ante un mieloma múltiple (MM) y ante la imposibilidad de completar el estudio para descartarlo y hacer el diagnóstico diferencial con otras GM desde Atención Primaria, se derivó a la paciente a las consultas de hematología donde le realizaron las pruebas complementarias necesarias para ello (tabla 1).

Tras el estudio se llegó al diagnóstico de GM de significado incierto (GMSI), ya que nos encontramos ante una paciente asintomática que presenta < 10% de células plasmáticas en la médula ósea (MO), proteína monoclonal < 3 g/dl y que no presenta lesiones osteolíticas en la serie ósea realizada. Esta entidad no precisa tratamiento, pero debido a la posibilidad de que evolucione hacia una GM maligna (GMM) es imprescindible un seguimiento clínico-biológico de por vida de esta paciente, por lo que fue citada para control en 6 meses.
DISCUSIÓN
La GMSI es una GM que se caracteriza por la presencia de una proteína monoclonal, estable en el tiempo, en ausencia de hallazgos que permitan el diagnóstico de MM o de otras GM con producción progresiva de proteína M1,3.
En la tabla 2 se expone la clasificación de las GM según el carácter transitorio, estable o progresivo de la producción de componente M1.

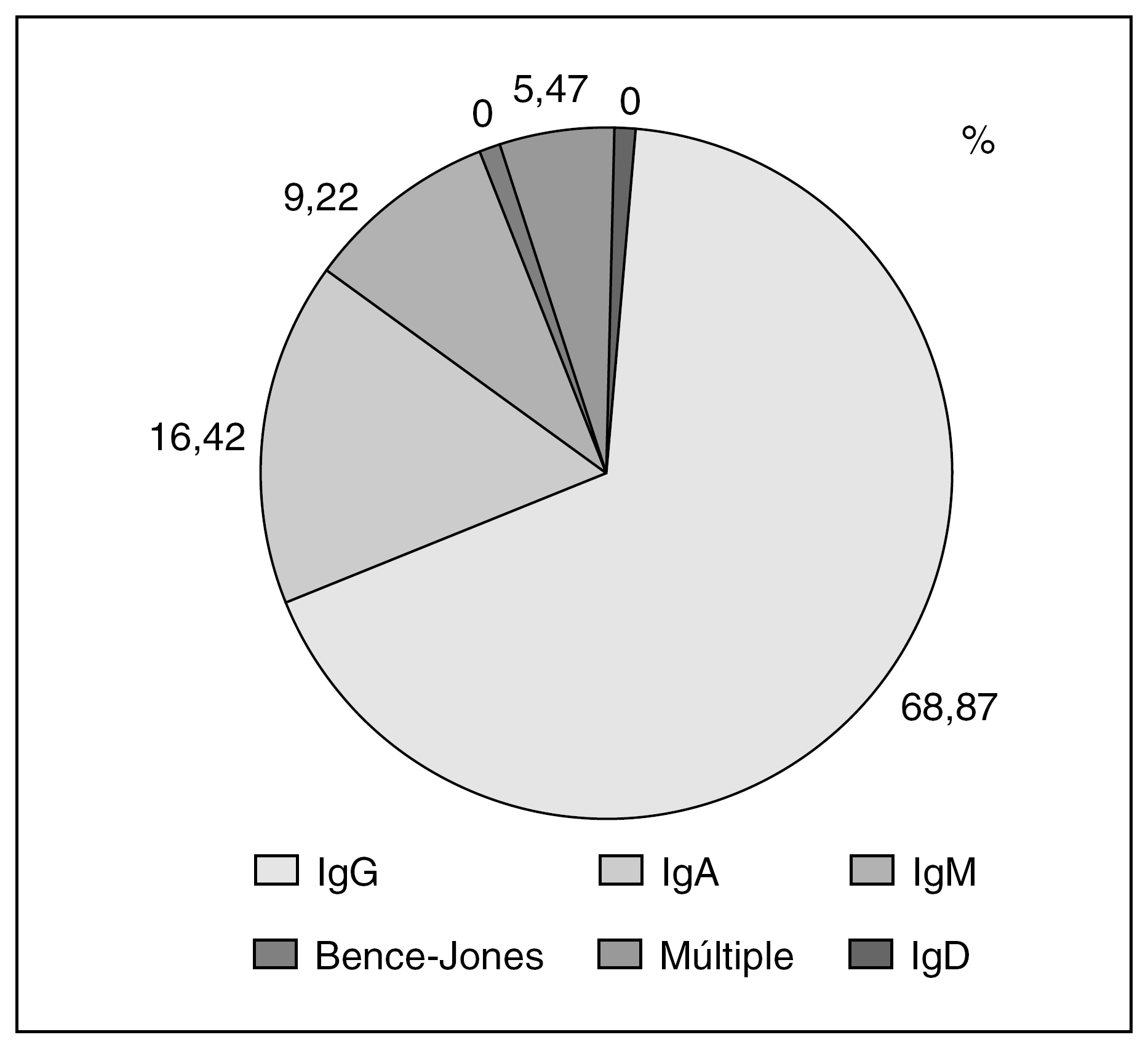
La incidencia de la GMSI aumenta con la edad y, así, entre el 1% y el 3% de las personas mayores de 70 años presentan esta alteración, proporción que aumenta cuando se utilizan técnicas más sensibles3. La edad media al momento del diagnóstico de la GMSI es de 62,1 años5. Respecto al tipo inmunohistoquímico la variedad IgG es la más frecuente5,6 (fig. 1).
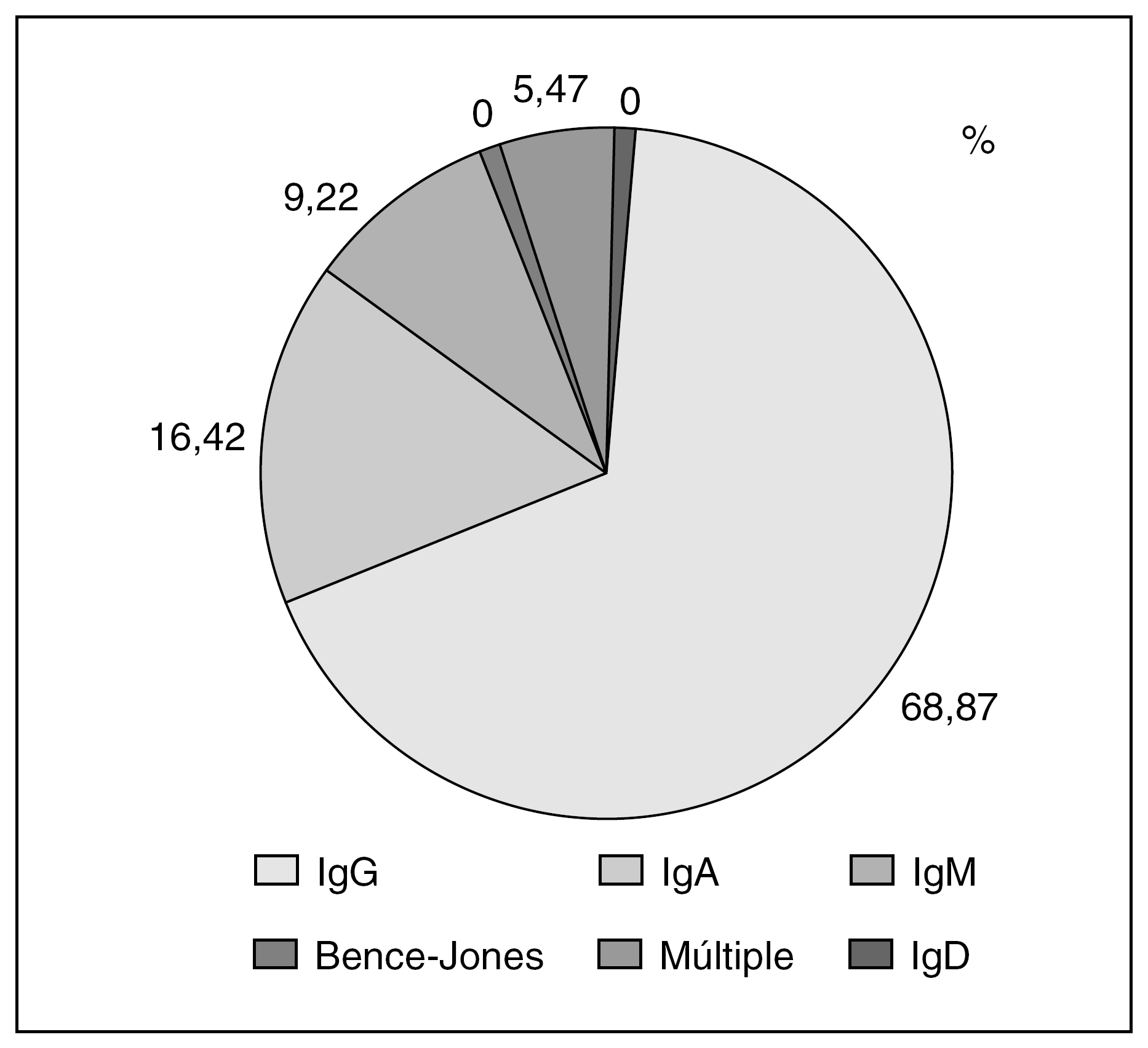
Figura 1. Tipos inmunoquímicos en la gammmapatía monoclonal de sistema incierto5. Ig: inmunoglobulinas.
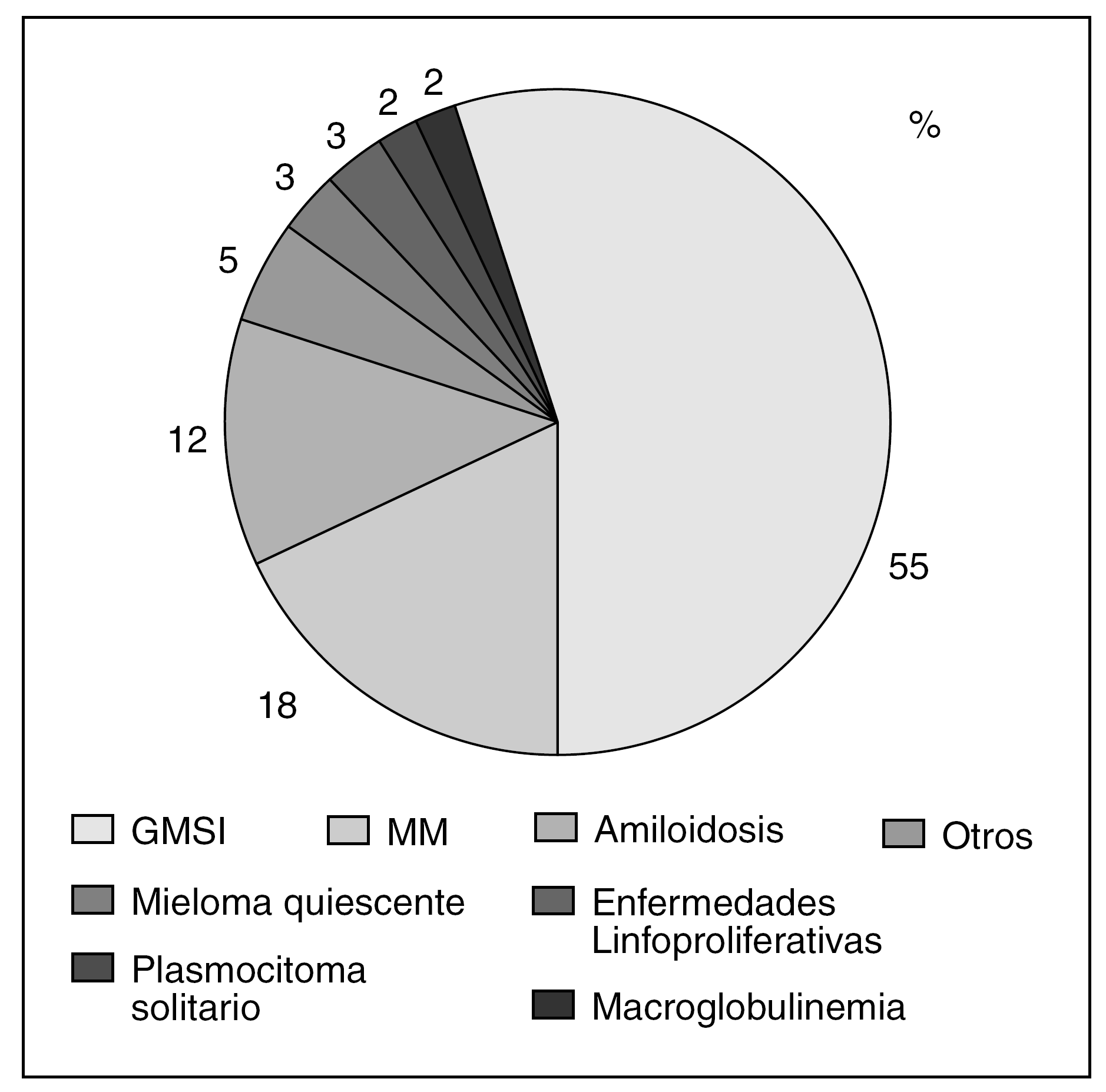
Aunque el MM constituye el prototipo de GM, la más frecuente, en la mayoría de las series, es la GMSI2,7. Así, en una serie de 882 pacientes con GM de la Clínica Mayo el 55% fueron diagnosticados de GMSI, mientras que el 18% lo fueron de MM8 (fig. 2); resultados similares obtuvo Kyle9 en una serie de 1.026 pacientes, en la que el 56% fue diagnosticado de GMSI y un 18% de MM. Sin embargo, Giraldo et al5, en una serie de 1.203 pacientes encontró una distribución equilibrada entre las dos patologías, el 33% fueron diagnosticados de MM y un 28,9% de GMSI.
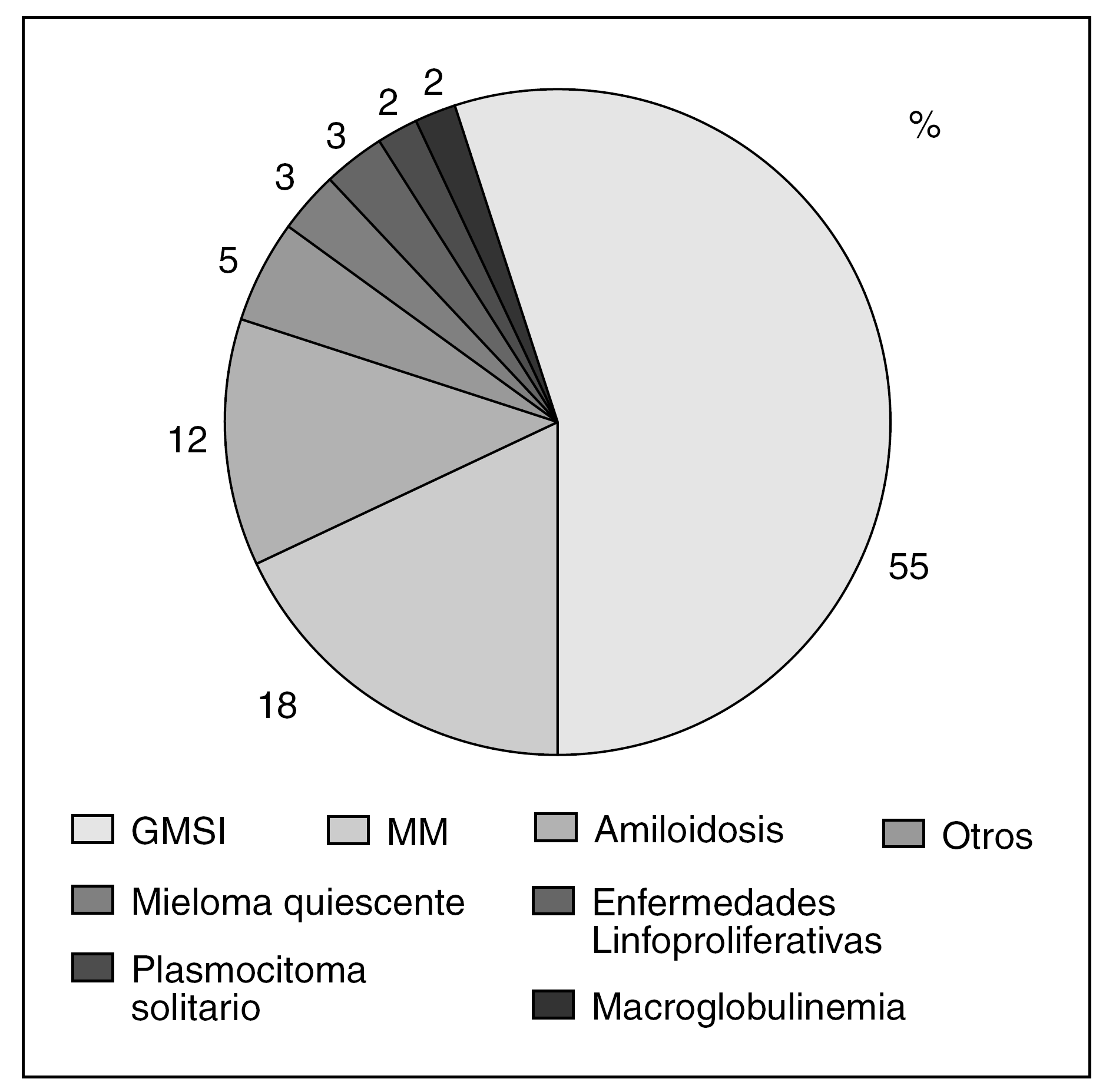
Figura 2. Distribución de las gammapatías monoclonales8. GMSI: gammmapatías monoclonales de sistema incierto; MM: mieloma múltiple.
Establecer el diagnóstico diferencial entre GMSI y MM suele ser sencillo y se basa generalmente en la aplicación de los criterios diagnósticos del MM, que incluyen la valoración de los parámetros clásicos: porcentaje de células plasmáticas medular, niveles del componente monoclonal en suero o en orina, y la presencia de lesiones osteolíticas. A éstos se han añadido otros factores complementarios presentes en el MM y que se consideran como de mal pronóstico o enfermedad avanzada, entre los que destacan anemia, hipercalcemia e insuficiencia renal5. Así, para establecer el diagnóstico definitivo de GMSI se exige demostrar valores de proteína M < 3 g/dl en sangre y ausencia o cantidades pequeñas en orina; menos de un 10% de células plasmáticas en MO; valores normales de creatinina, calcio y hemoglobina; ausencia de lesiones líticas y valores estables del componente M en el tiempo2,10. Naturalmente, para llegar a establecer el diagnóstico se precisa un estudio que incluya una punción de MO, la cual no puede ser realizada en Atención Primaria, por lo que es necesario derivar al paciente al hematólogo. Aunque este diagnóstico diferencial suele ser sencillo, existe un pequeño número de pacientes asintomáticos en los que es difícil hacerlo. Para ellos se ha acuñado el término de mieloma quiescente (smoldering myeloma), éstos pacientes presentan un componente M > 3 mg/dl y > 10% de células plasmáticas en MO, en ausencia de lesiones osteolíticas y sintomatología clínica1 (tabla 3).

La importancia del conocimiento de estas dos entidades reside en que en ellas, a diferencia del MM, la quimioterapia no es eficaz y no precisan tratamiento. Aunque en un estudio en el que se comparó la supervivencia de una población de su misma edad y sexo la presencia de GMSI no se asociaba de forma significativa a un acortamiento de la supervivencia11, no hay que olvidar que existe la posibilidad de evolución de la GMSI hacia GMM1,5. El mieloma quiescente se comporta de forma similar, aunque el mayor grado de infiltración medular y la magnitud del componente M deriva en una evolución más frecuente a discrasias malignas de células plasmáticas12.
Kyle13 estudió la evolución de la GMSI en un amplia serie de 241 pacientes. Observó que a los 5 años tan sólo el 11% de ellos habían evolucionado a MM, macroglobulinemia o amiloidosis, mientras que el 57% mantenían valores estables del componente M, el 9% los habían incrementando por lo menos un 50%, pero permanecían asintomáticos, y el 23% fallecieron por causas no relacionadas. A los 10 años el porcentaje de malignización alcanzaba el 18%, el 33% a los 20 años y el 40% a los 25 años14. Resultados que vienen a coincidir con otros autores3,5,11,15,16. Aunque la mediana del intervalo desde el diagnóstico de la GMSI hasta la aparición de GMM se sitúa en 10 años (2-29 años)17, es importante señalar que la forma de transición hacia GMM puede ser variable: con un incremento lento pero progresivo del componente M desde el diagnóstico de la GMSI; con aumento progresivo del componente M hasta el diagnóstico de una GMM en pacientes que durante años habían mantenido concentraciones estables; o con elevaciones bruscas de la concentración del componente M en pacientes previamente estables3,17.
Aunque se han intentado relacionar diferentes parámetros clínicos y analíticos con mayor probabilidad de evolución de la GMSI a GMM no existen parámetros fiables para predecir la transformación maligna de una GMSI, siendo solamente el seguimiento clínico-biológico periódico lo que nos permite valorar la evolución de estos pacientes. En este sentido, un estudio de Bladé et al18 indica que dicha probabilidad sería superior en las GMSI de tipo IgA, aunque el número de personas con este tipo de gammapatía es escaso; y en una reciente serie de Kyle et al19 en 1.384 pacientes con GMSI, la concentración y el tipo de Ig fueron el único factor independiente de progresión, mientras que la presencia de proteína monoclonal en orina y la disminución de una o varias Ig no fueron factores de progresión.
No se ha referido una mayor agresividad clínica de los MM que siguen a GMSI3. En la serie de Kyle la mediana de supervivencia desde el diagnóstico de MM fue de 34 meses17.
La imposibilidad de definir un grupo de GMSI de mayor riesgo de evolución a MM, la presentación abrupta de éste en muchos casos y la gran variabilidad en el intervalo entre la detección de la GMSI (en varios casos la transformación maligna se ha producido más allá de los 25 años del diagnóstico de la GM) y el diagnóstico del MM, hacen necesario un seguimiento periódico e indefinido de todas las personas con GMSI. La frecuencia de esta monitorización no está claramente determinada, aunque el control clínico y analítico, primero semestral y luego anual, de estos pacientes parece suficiente20. En el seguimiento de estos pacientes debe prestarse especial interés al estudio minucioso de la sintomatología ósea de nueva aparición, así como a la monitorización de determinados parámetros bioquímicos, como hemograma con VSG, proteinograma y proteinuria, calcemia y creatinina.
Correspondencia: B. de Rivas
Escosura, 6 - 6.o A
28015 Madrid
Correo electrónico: rivasotero@yahoo.es
Recibido el 2-5-2004; aceptado para su publicación 18-10-2004.
