




INTRODUCCIÓN
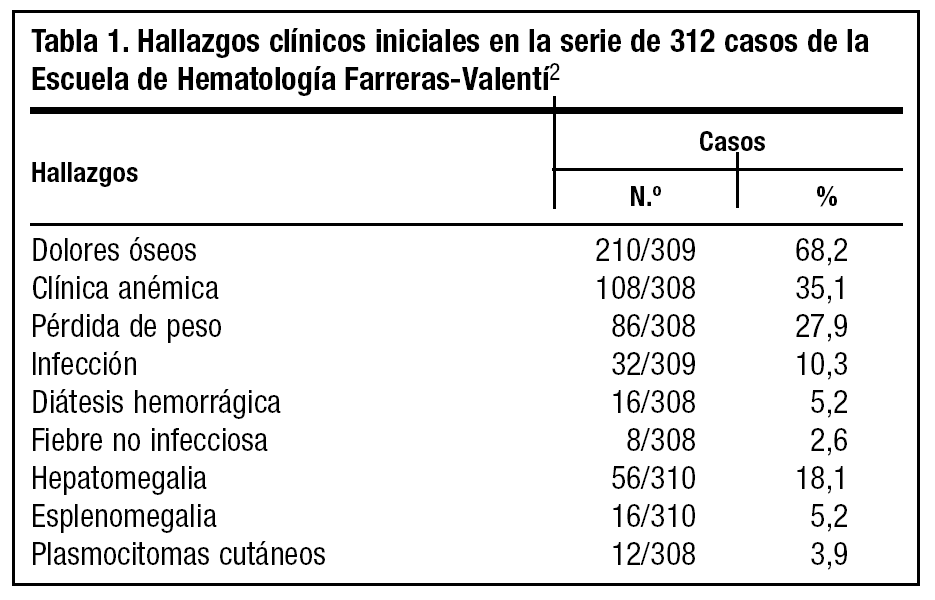
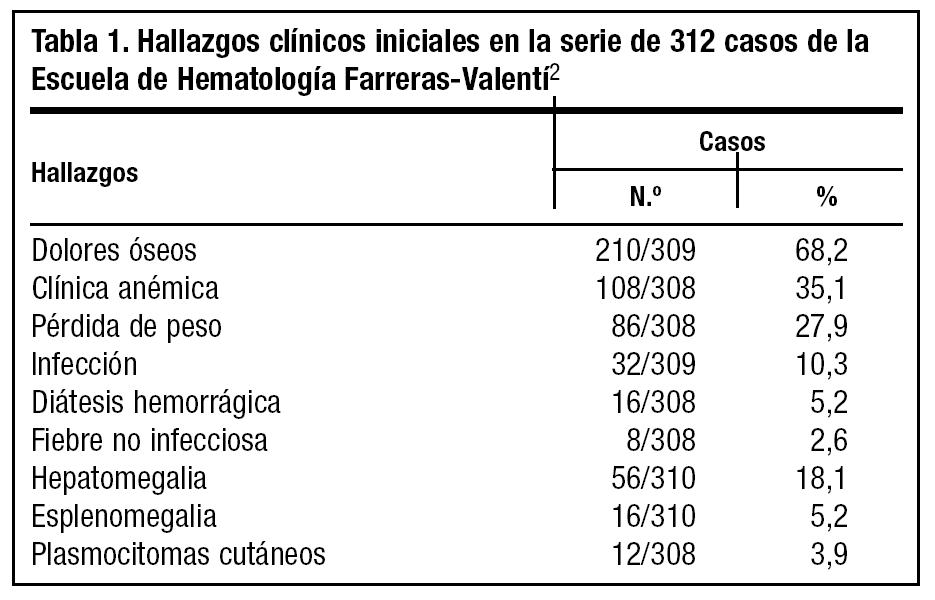
El mieloma múltiple es el prototipo de gammapatía monoclonal maligna. Las manifestaciones se deben a la proliferación tumoral plasmocelular (lesiones esqueléticas, anemia, hipercalcemia e infiltración de diversos órganos y tejidos) y a la producción de la proteína monoclonal por parte de células mielomatosas (insuficiencia renal, predisposición a infecciones, síndrome de hiperviscosidad)1. La edad media se sitúa en 60-65 años, sólo entre un 15% y un 2% de los pacientes tienen menos de 50 y 40 años, respectivamente. Los dolores óseos constituyen el síntoma más frecuente. El dolor se localiza preferentemente en la columna vertebral y la parrilla costal, presenta características mecánicas y se exacerba con los movimientos y con la tos. La anemia también es una manifestación clínica frecuente. Puede existir afectación del estado general, con astenia y pérdida de peso. La fiebre debida a la propia enfermedad se da en menos del 5% de los enfermos2. En muchos casos la primera manifestación la constituyen infecciones de repetición, entre las que destaca la neumonía neumocócica. Otras veces la enfermedad se manifiesta con insuficiencia renal o con sintomatología secundaria a hipercalcemia (náuseas, vómitos, poliuria, polidipsia, estreñimiento, cefaleas, somnolencia, irritabilidad e incluso coma). La diátesis hemorrágica, en forma de epistaxis, hematurias o equimosis, puede ser también manifestación inicial (tabla 1).
En el 90% de los pacientes la velocidad de sedimentación globular (VSG) está muy aumentada. El 60-70% presenta anemia por infiltración glomerular, siendo el recuento de leucocitos casi siempre normal. Una cuarta parte de los pacientes presenta insuficiencia renal. En el 90% de los mielomas de inmunoglobulina (Ig)G e IgA la viscosidad plasmática es superior a la normal. El aspirado de médula ósea suele mostrar una infiltración por células plasmáticas superior al 20%3.
Pronóstico
La supervivencia media de los pacientes es, en general, de 2-3 años, variando mucho de unos enfermos a otros2. Los principales factores pronósticos son4:
Insuficiencia renal (creatinina > 2 mg/dl).
Anemia (hemoglobina [Hb] > 85 g/l).
Hipercalcemia (calcio sérico > 11,5 mg/dl o > 2,86 mmol/l).
Hipoalbuminemia (albúmina < 40 g/l).
Trombocitopenia (recuento de plaquetas < 100 x 10 elevado a 9 l).
Morfología plasmoblástica.
Tipo Bence-Jones lambda o IgD.
Destrucción esquelética intensa.
Beta2 macroglobulina sérica > 6 mg/l.
Índice proliferativo elevado.
Respuesta muy rápida al tratamiento (< 2 meses).
Ausencia de respuesta al tratamiento (progresión de la enfermedad).
La insuficiencia renal es el factor individual con mayor influencia pronóstica desfavorable. No obstante, el factor pronóstico más importante es la sensibilidad de la clona mielomatosa al tratamiento citostático. Con la quimioterapia se alcanza una respuesta objetiva en alrededor del 50% de los pacientes que persiste 1-2 años, al cabo de los cuales suele producirse una recaída. La respuesta al tratamiento es cada vez menos duradera. La infección constituye la causa más frecuente de muerte5.
La mayoría de los pacientes con mieloma múltiple tienen síntomas o alteraciones analíticas en el momento del diagnóstico, que indican enfermedad activa y que requieren tratamiento citostático3. A nuestro paciente se le administró por parte del Servicio de Hematología adriamicina, vincristina y dexametasona durante 4 días, con buena tolerancia y sin efectos secundarios.
El melfalán y la ciclofosfamida, aislados o combinados con prednisona, han sido los fármacos más empleados en el tratamiento1. Si existe trombocitopenia o insuficiencia renal es preferible administrar ciclofosfamida que melfalán. Sin embargo, la proporción de pacientes que viven 5 años o más es sólo del 10-20%, por lo que se están empleando pautas poliquimioterápicas como VCMP (vincristina, ciclofosfamida, melfalán y prednisona), VBAP (vincristina, BCNU [biscloroetil nitrosourea], adriamicina y prednisona), VCAP (vincristina, ciclofosfamida, adriamicina y prednisona), obteniéndose un mayor número de respuestas objetivas.
EXPOSICIÓN DEL CASO
Varón de 40 años sin antecedentes personales de interés ni hábitos tóxicos ni yatrogenia, que acude a consulta refiriendo pérdida de peso de aproximadamente 12 kg los últimos 2-3 meses, junto con astenia, poliuria, polidipsia y nicturia de 2-3 veces. A la exploración física no notamos nada significativo salvo la constatación de pérdida de peso ya referida. En la analítica general destacan: Hb 13,3 g/dl, volumen corpuscular medio (VCM) 94,1 mm3, hematocrito 39,9%, VSG 104/116 mm/h, Ácido úrico 7,9 mg/dl, fosfatasa alcalina 244 U/l, gammaglutamiltranspeptidasa (GGT) 476 U/l, creatinina 5,97 mg/dl, potasio 5,3 meq/l, calcio 9,5 mg/dl, resto normal. Al paciente se le realiza ecografíaabdominal: hígado, vesícula, vía biliar y páncreas sin alteraciones. Esplenomegalia. Riñones de tamaño ligeramente aumentado, con aumento de ecogenicidad cortical con pirámides hipoecoicas. Se le realiza proteinograma apreciándose pico beta con elevación de beta2 microglobulina, así como punción de médula ósea: plasmocitosis compatible con mieloma múltiple.
DISCUSIÓN
Se trata de un debut con síndrome constitucional, aumento GGT y fosfatasa alcalina y deterioro grave del filtrado glomerular, en un varón joven (40 años), constituyendo una presentación relativamente atípica de la enfermedad. El riñón se afecta en aproximadamente la mitad de los pacientes con mieloma múltiple. El 25-30% tienen insuficiencia renal en el momento del diagnóstico y en el resto aparece en el curso de la enfermedad (tabla 1). La insuficiencia renal es el factor individual con mayor influencia pronóstica desfavorable1. No obstante, el factor pronóstico más importante es la sensibilidad de la clona mielomatosa al tratamiento citostático. Con la quimioterapia se alcanza una respuesta objetiva en alrededor del 50% de los pacientes que persiste 1-2 años, al cabo de los cuales suele producirse una recaída. La infección constituye la causa más frecuente de muerte. La mayoría de los pacientes tienen síntomas o alteraciones analíticas en el momento del diagnóstico, que indican enfermedad activa y que requieren tratamiento citostático. El melfalán y la ciclofosfamida, aislados o combinados con prednisona, son los fármacos más empleados en el tratamiento1. Sin embargo, la proporción de pacientes que viven 5 años o más es sólo del 10-20%. La evolución de la enfermedad en pacientes jóvenes suele ser lenta y la supervivencia prolongada, aunque se han descrito casos de mieloma en pacientes jóve-nes con comportamiento clínico agresivo y supervivencia corta.
Correspondencia: S. Santamaría Carmona.
C/ El Toboso, n.º 1.
18199 Cájar. Granada.
Correo electrónico: soniasc@latinmail.com
Recibido el 21-10-04; aceptado para su publicación el 21-02-05.
