





La histiocitosis pulmonar de células de Langerhans es una enfermedad rara de adultos jóvenes, generalmente fumadores, que se asocia con una significativa morbilidad. El curso de esta enfermedad es impredecible, oscilando desde formas benignas autolimitadas hasta formas malignas con evolución progresiva hacia la insuficiencia respiratoria y la muerte. Su causa es desconocida, pero la mayor parte de los autores creen que en estos pacientes hay una alteración en la regulación del sistema inmunológico. Presentamos el caso de un paciente con el diagnóstico de histiocitosis pulmonar de células de Langerhans y revisamos las características clínicas, diagnósticas y terapéuticas de esta enfermedad. Consideramos de interés este caso por la baja frecuencia de esta entidad.
Pulmonary Langerhans cell histiocytosis is a rare disease of young adults, usually smokers, which is associated with significant morbidity. The course of the disease is unpredictable, ranging from benign self-limiting types with spontaneous regression, to malignant forms with progression to respiratory failure and death. Its cause is unknown, but most authors believe that there is an alteration in immune system regulation in these patients. We report a case of a patient with the diagnosis of pulmonary Langerhans cell histiocytosis and review the clinical characteristics, diagnosis and treatment of this disease. We consider this case of interest due by the low frequency of this disease.
La histiocitosis de células de Langerhans (HCL) es una enfermedad rara que se caracteriza por la infiltración de uno o más órganos por células de Langerhans (células de la línea monocitos-macrófagos que presentan un núcleo grande e inclusiones citoplásmicas denominadas gránulos de Birbeck)1. La forma localizada de HCL se denominaba anteriormente granuloma eosinófilo, mientras que las variantes multisistémicas se conocían con los nombres de enfermedad de Letterer-Siwe y enfermedad de Hand-Schüller-Christian. En 1953, Lichtenstein habló por primera vez de histiocitosis X para referirse a estas 3 entidades. Actualmente, el Writing Group de la Histiocyte Society recomienda sustituir este término por histiocitosis de células de Langerhans2. El término histiocitosis pulmonar de células de Langerhans (HPCL) se usa para referirse a la enfermedad cuando cursa con afectación pulmonar, ya sea de forma aislada o englobando además a otros órganos. Para evitar confusiones, se ha establecido un sistema de clasificación simplificado (tabla 1).
Sistema simplificado de clasificación de las histiocitosis de células de Langerhans en adultos
| Afectación de un solo órgano |
| Pulmón (afectación única en más del 85% de los casos que presentan afectación pulmonar) |
| Hueso |
| Piel |
| Hipófisis |
| Ganglios linfáticos |
| Otras localizaciones: tiroides, hígado, bazo, sistema nervioso central |
| Enfermedad multisistémica |
| Enfermedad multisistémica con afectación pulmonar |
| Enfermedad multisistémica sin afectación pulmonar |
| Enfermedad histiocítica de múltiples órganos |
Tomada de García et al.2.
Varón de 44 años de edad que consultó inicialmente por tos productiva matutina (expectoración blanquecina), astenia progresiva y disnea de grandes esfuerzos, de 8 meses de evolución. Entre los antecedentes destacaba ser fumador activo de al menos 40 cigarrillos al día, desde hacía más de 25 años. No refería infecciones respiratorias de repetición, no había viajado recientemente al extranjero ni había tenido contacto con animales. No tomaba drogas ni medicamentos. Negaba exposición a tóxicos ambientales, siendo su profesión auxiliar administrativo. No había dolor torácico, ni hemoptisis ni fiebre. No tenía dolores óseos. En la exploración física, el paciente presentaba una buena coloración e hidratación adecuada. Las constantes vitales estaban dentro de la normalidad. No se palpaban adenopatías a ningún nivel y la auscultación pulmonar no mostró alteraciones significativas. El resto de la exploración física era rigurosamente normal.
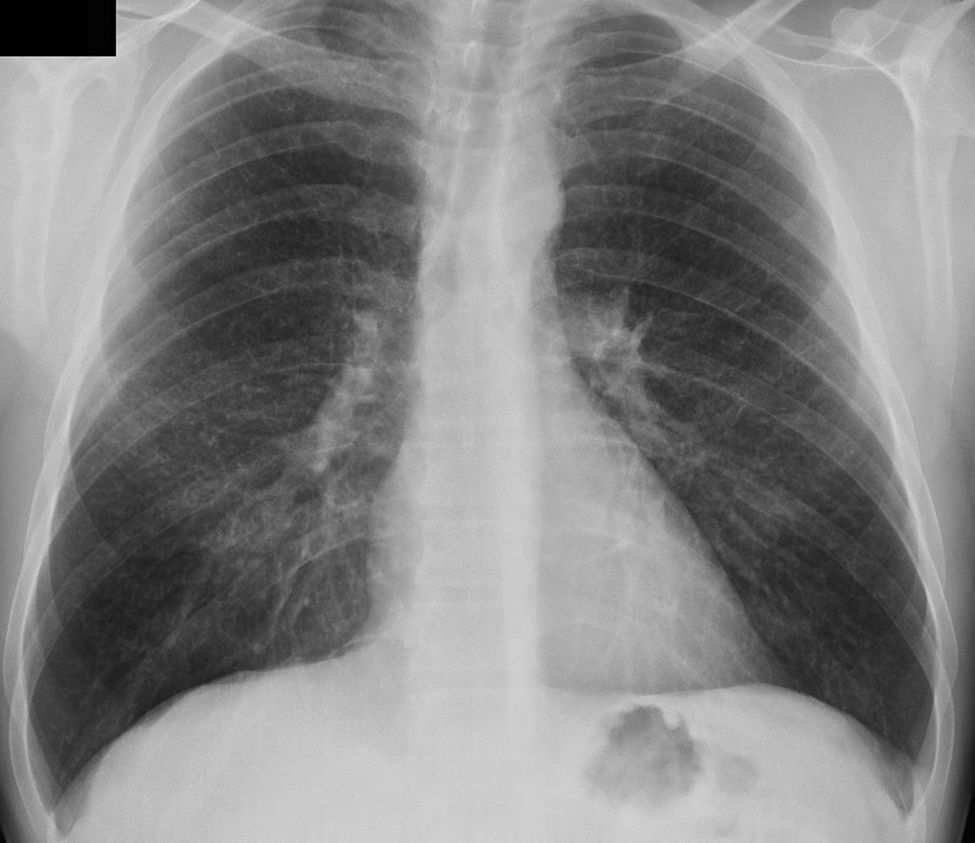
Se solicitó una radiografía pulmonar, donde se apreció un patrón intersticial de predominio micronodular bilateral, en campos medios y superiores (fig. 1). Se realizó analítica de sangre que incluyó hemograma, bioquímica, coagulación, estudio de autoinmunidad y factor reumatoide, siendo todos los resultados normales. La prueba de Mantoux fue negativa. Ante estos hallazgos se remitió al paciente a neumología para completar el estudio.
.")
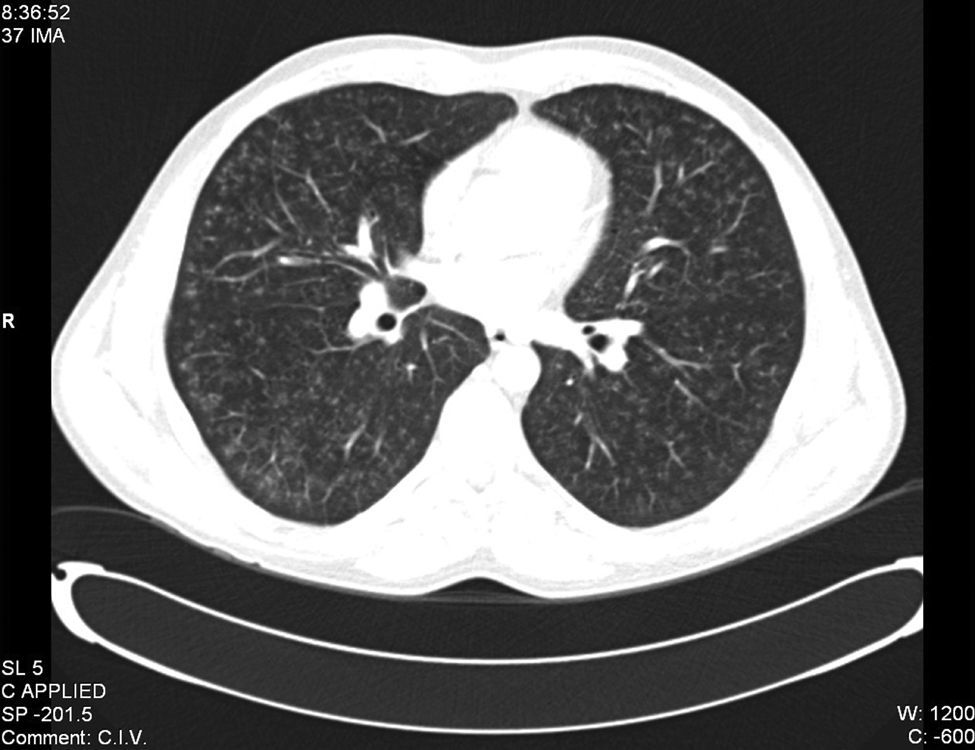
Se realizaron pruebas funcionales respiratorias que mostraron un patrón normal, con disminución moderada de la difusión. El ecocardiograma transtorácico mostró insuficiencia tricuspídea ligera, descartándose hipertensión pulmonar asociada. En la tomografía computarizada (TC) torácica se apreció patrón intersticial, caracterizado por lesiones micronodulares y microquísticas, fundamentalmente en campos medios y superiores (fig. 2). Se hizo una videobroncoscopia donde se visualizaron, en ambos sistemas bronquiales, signos de bronquitis y una mucosa globalmente edematosa, con aumento de la vascularización, sobre todo en las vías proximales. No se evidenciaron lesiones endobronquiales circunscritas ni signos de sangrado. Se realizó lavado broncoalveolar a través de la língula que se procesó para citobioquímica y citología. Los resultados posbroncoscopia mostraron negatividad para bacilos ácido-alcohol resistentes y el cultivo de Lowenstein también resultó negativo. La citología fue negativa para malignidad, evidenciando células epiteliales sin alteraciones citológicas relevantes, con un 98% de macrófagos, un 1% de linfocitos y un 1% de neutrófilos. No se realizó inmunohistoquímica para células CD1 por un error de laboratorio. La anatomía patológica de la muestra obtenida por biopsia transbronquial mostró un parénquima pulmonar con patrón de fibrosis intersticial crónica. Finalmente se realizó una biopsia pulmonar mediante toracoscopia. El estudio microscópico reveló la presencia de lesiones difusas de carácter parcheado, en las que se observaba infiltración por eosinófilos y células histiocitarias de citoplasma acidófilo y núcleo hendido, sobre todo en las paredes alveolares y alrededor de los vasos y bronquios. También se encontraron áreas de fibrosis. Con el diagnóstico de histiocitosis pulmonar de células de Langerhans se recomendó al paciente el abandono del tabaco como principal estrategia para tratar la enfermedad.
.")
Tomografía computarizada. Patrón intersticial, caracterizado por lesiones micronodulares y microquísticas. Hallazgos presentes fundamentalmente en campos pulmonares medios y superiores.
Fuente: elaboración propia, imagen aportada por el servicio de radiodiagnóstico del Hospital Infanta Cristina de Parla (Madrid).
La HCL en una enfermedad rara (en un estudio realizado en Suiza entre 1970 y 1991 se encontraron únicamente 19 casos)3. Su causa es desconocida, aunque la mayor parte de los investigadores sostienen que los pacientes presentan una disfunción del sistema inmunológico. Otros autores señalan que podría tratarse de un proceso neoplásico o que estarían implicados virus o factores genéticos4. La enfermedad se ha relacionado con la espondilitis anquilosante5. Más del 90% de los pacientes con HCL tienen historia de tabaquismo y la enfermedad suele presentarse entre los 30 y los 40 años de edad6,7. No está claro si hay diferencias por sexo: las diferencias que se encuentran en los distintos estudios podrían ser reflejo de las diferencias existentes entre sexos en lo que se refiere al hábito de fumar. No existen asociaciones familiares y la enfermedad se da principalmente en la raza blanca1,2. Su incidencia y prevalencia no se conocen con certeza, aunque en estudios de biopsias pulmonares en pacientes con enfermedad intersticial se encuentran datos de HPCL en un 5% de los casos, lo que indica que es una entidad rara2,6.
El 25% de los pacientes no presentan síntomas en el momento de diagnóstico y en muchas ocasiones la enfermedad se diagnostica de forma casual al hacer una radiografía torácica de rutina1,2. Clínicamente, los pacientes con HPCL pueden presentar disnea, tos o dolor torácico. Otros síntomas menos frecuentes son hemoptisis y síndrome constitucional y se han descrito pacientes con neumotórax espontáneo. En algunos casos se pueden encontrar síntomas por afectación de otros órganos: diabetes insípida, dolor abdominal, adenopatías, afectación cutánea o lesiones óseas. La enfermedad se ha asociado con neoplasias como el linfoma de Hodgkin o no hodgkiniano, el ganglioneuroma mediastínico y el carcinoma broncogénico8. Es probable que el efecto carcinogénico del tabaco explique en parte esta asociación.
La exploración física, como ocurre en nuestro caso, no suele mostrar hallazgos significativos2,7,9. Según el grado existente de fibrosis pulmonar, sí se pueden encontrar alteraciones en la auscultación pulmonar2,7.
Los exámenes de laboratorio son inespecíficos. Las pruebas de función pulmonar pueden estar alteradas, presentando un patrón obstructivo, restrictivo o mixto. Los volúmenes pulmonares son normales o están incrementados y lo más característico es la disminución en la capacidad de difusión del monóxido de carbono, como ocurre con nuestro paciente (se puede encontrar esta alteración en al menos el 70% de los casos)7. La radiografía de tórax es anormal en la mayor parte de los pacientes1. Suelen aparecer micronódulos o retículo-nódulos y afectación intersticial, con lesiones generalmente bilaterales y simétricas. Estas alteraciones se suelen presentar sobre todo en lóbulos superiores y medios y tienden a respetar los ángulos costofrénicos6,7. La TC de tórax es una herramienta útil y sensible para el diagnóstico. En la evolución de la enfermedad se pueden encontrar nódulos, nódulos cavitados, quistes de paredes gruesas y, finalmente, quistes de paredes delgadas. La coexistencia en la TC de nódulos, nódulos cavitados y quistes, es muy indicativa de histiocitosis pulmonar de células de Langerhans6. El hallazgo más frecuente en la HPCL son los quistes10. En nuestro caso, los hallazgos radiológicos fueron compatibles con lo descrito en la literatura médica para la HPCL. La confirmación del diagnóstico puede hacerse mediante lavado broncoalveolar, biopsia transbronquial o biopsia quirúrgica. Un número aumentado de células de Langerhans en el lavado broncoalveolar, identificadas mediante tinción con anticuerpos frente a CD1a, es muy indicativo de HPCL (cuando la proporción es ≥ 5% el diagnóstico es muy probable1). La broncoscopia con toma de biopsia transbronquial tiene un bajo rendimiento por la distribución parcheada de las lesiones y la escasa cantidad de tejido que se obtiene. La biopsia quirúrgica abierta o mediante toracoscopia es la prueba con mayor rendimiento diagnóstico, aunque también tiene falsos negativos y su indicación debe ser individualizada, ya que supone riesgos para el paciente. En el diagnóstico diferencial, principalmente hay que tener en cuenta sarcoidosis, silicosis, linfangioleiomiomatosis y alveolitis alérgica extrínseca1.
El principal objetivo terapéutico debe ser siempre el abandono del consumo de tabaco1,6,7. Dada la frecuencia de remisiones espontáneas, debe mantenerse al paciente en observación si está asintomático, incidiendo en el abandono de este hábito. Si está sintomático o la enfermedad progresa, se puede iniciar tratamiento con corticoides durante 6-12 meses (por ejemplo, 0,5-1mg/kg/día de prednisona)6. Si no hubiera respuesta, se puede recurrir a los inmunosupresores como el etopóxido, metotrexato, ciclofosfamida o vinblastina. En fases muy evolucionadas, puede ser necesario el trasplante pulmonar, aunque se han descrito recurrencias de la enfermedad en el pulmón trasplantado7. El curso de la enfermedad es variable y su espectro va desde pacientes con remisión espontánea tras dejar de fumar, hasta la enfermedad progresiva y la muerte. Son factores de mal pronóstico las edades extremas, las formas que cursan con afectación multiorgánica o con síndrome constitucional mantenido, neumotórax de repetición, un elevado número de quistes e imagen en panal de abeja en la radiografía de tórax, una marcada disminución de la capacidad de difusión, la hipertensión pulmonar asociada, la administración prolongada de corticoides, la alta proporción de volumen residual respecto a la capacidad pulmonar total o una baja proporción de volumen espiratorio forzado en un segundo en relación con la capacidad vital forzada1,6,7.
Tras abandonar el consumo de tabaco, nuestro paciente ha presentado gran mejoría clínica y está pendiente de nuevas pruebas de imagen que confirmen la mejora radiológica, su pronóstico es favorable.
La HPCL es una entidad rara y de diagnóstico difícil, porque puede cursar de forma asintomática, porque muchos pacientes presentan remisión espontánea y porque la biopsia pulmonar puede no ser concluyente. Hemos considerado interesante la exposición de este caso de HPCL, ya que creemos que su descripción ilustra los hallazgos clínicos, radiológicos e histopatológicos que se pueden encontrar en esta enigmática enfermedad.
Responsabilidades ÉticasProtección de personas y animales. Los autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datos. Los autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informado. Los autores declaran que en este artículo no aparecen datos de pacientes.
