




INTRODUCCIÓN
La farmacovigilancia puede definirse como el estudio de la seguridad de los fármacos en las condiciones de uso de la práctica clínica en grandes comunidades1. Si bien otras definiciones amplían el ámbito de la farmacovigilancia e incluyen, además, el desarrollo clínico y preclínico de los fármacos2, la farmacovigilancia es, en rigor, una actividad de salud pública destinada a analizar y gestionar los riesgos de los medicamentos una vez comercializados3.
La mayor importancia de la farmacovigilancia radica en el hecho de que el que se haya registrado un nuevo medicamento no significa que se conozca todo sobre el mismo: los ensayos clínicos precomercialización se realizan en un número de pacientes que resulta insuficiente para detectar una reacción adversa poco frecuente: basta pensar que para detectar tres casos de una reacción adversa cuya incidencia sea del 0,1% son necesarios 6.500 pacientes, mientras que este número se incrementa a 65.000 pacientes en el caso de que la incidencia de la reacción adversa sea del 0,01%4. Además, los ensayos clínicos precomercialización se realizan en unas condiciones muy estandarizadas, que limitan la extrapolación de los resultados a las condiciones habituales de uso. Así, en la práctica habitual, el número de pacientes expuestos al tratamiento es mucho mayor que en los ensayos clínicos, la duración del tratamiento puede ser más larga, pueden tratarse poblaciones especiales no estudiadas de forma específica durante el desarrollo clínico, la patología concomitante es más frecuente, la posibilidad de interacciones es mayor y el cumplimiento terapéutico no se controla del mismo modo4. Por dichas razones está claro que, al prescribir un tratamiento farmacológico, el médico puede enfrentarse en la práctica diaria a situaciones nuevas y distintas de aquéllas reflejadas previamente en la literatura y puede observar y/o sospechar reacciones adversas hasta el momento desconocidas o que, aun siendo conocidas, impliquen un aumento de la incidencia y/o de la gravedad, lo cual aporta un conocimiento sin duda importante. En este sentido debe tenerse en cuenta que algunos fármacos han visto restringida su utilización o han sido retirados debido a reacciones adversas graves. Basta recordar los ejemplos de: tocainida (neutropenia), benoxaprofeno (hepatotoxicidad, reacciones cutáneas graves), flecainida (arritmias), xamoterol (empeoramiento de la insuficiencia cardíaca), trovafloxacino (hepatotoxicidad), troglitazona (hepatotoxicidad), terodilina (arritmias), fenfluramina y dexfenfluramina (trastorno valvular) y sertindol (arritmias) y cerivastatina (rabdomiólisis), entre otros1,5, así como la reciente retirada del mercado de rofecoxib debido al riesgo de accidentes cardiovasculares graves6. Es innegable, por tanto, la relevancia del médico en la detección y comunicación de reacciones adversas, lo que contribuirá a un conocimiento más preciso del perfil de toxicidad de los fármacos en las condiciones de uso habitual.
En el presente trabajo intentamos explicar la implicación de los clínicos, a nivel médico y legal, en materia de farmacovigilancia, así como proporcionar algunas definiciones de términos que pueden ser útiles a la hora de evaluar y notificar una sospecha de reacción adversa. También se menciona el tipo de información que deben proporcionar, si ello es posible, junto con algunas directrices útiles para evaluar la relación de causalidad. Por último, se exponen algunos procedimientos cuantitativos de generación de señales y algunos conceptos de gestión del riesgo en farmacovigilancia, todo ello con el objetivo de concienciar a los médicos de la importancia de la notificación espontánea de reacciones adversas y de clarificar, de alguna forma, el "camino" que siguen dichas notificaciones hasta la generación y/o confirmación de hipótesis de posibles riesgos asociados a medicamentos.
IMPLICACIÓN DE LOS MÉDICOS EN MATERIA DE FARMACOVIGILANCIA
Implicaciones a nivel médico
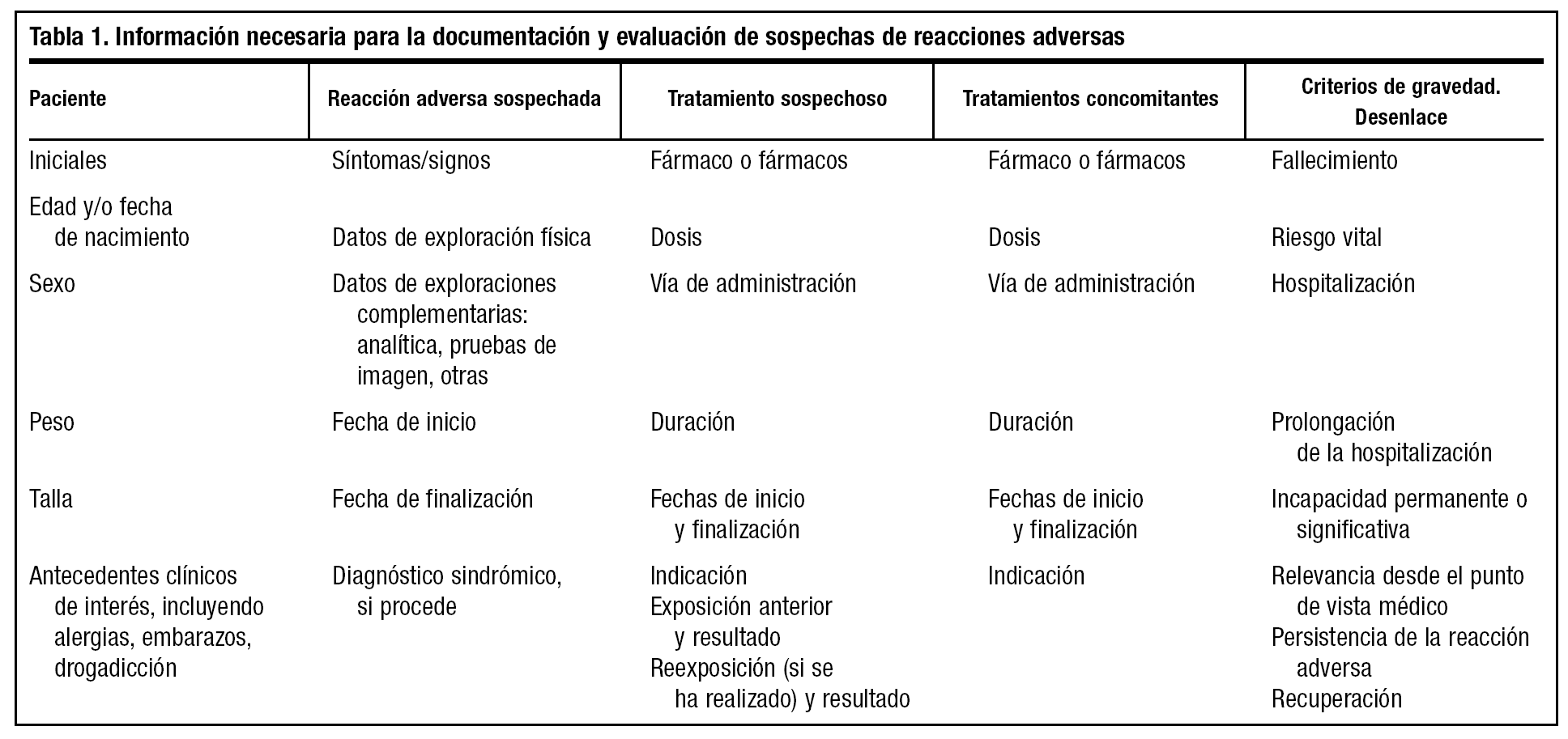
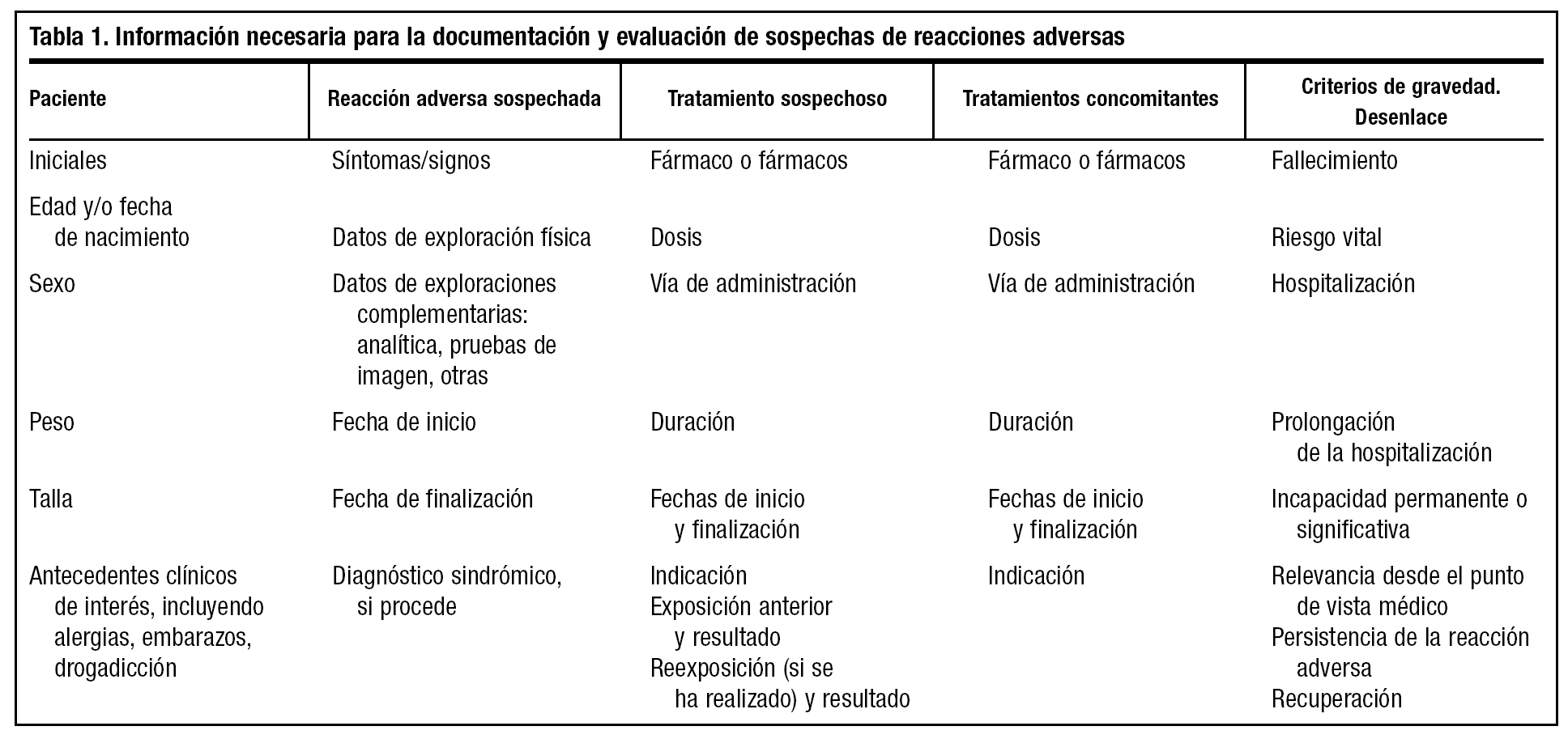
El médico posee un papel primordial en la detección de reacciones adversas a fármacos, principalmente mediante la notificación de los casos en los cuales sospeche que un fármaco puede haber producido una reacción adversa. El médico no debe dejar de comunicar una sospecha de reacción adversa porque no esté completamente seguro de que se deba al fármaco sospechoso. Toda información puede ser útil. Además, en nuestra opinión, ante la sospecha de una reacción adversa es bueno que el médico lo notifique a la compañía titular de la autorización de comercialización, contactando con el responsable de farmacovigilancia o a través del delegado de visita médica que, a su vez, informará al responsable de farmacovigilancia de la compañía, para el cual será muy útil recoger toda la información necesaria del clínico que ha visto al paciente y que se resume en la tabla 1. Para la correcta documentación de las notificaciones de sospechas de reacciones adversas es importante el conocimiento de un lenguaje estandarizado en cuanto a definiciones comúnmente utilizadas en farmacovigilancia, así como de los argumentos que se emplean en el establecimiento de la relación de causalidad. Ambos aspectos se tratarán más adelante.
Implicaciones a nivel legal
La responsabilidad de los médicos en materia de farmacovigilancia se halla regulada en el Real Decreto 711/2002, de 19 de julio7, donde se especifica que los profesionales sanitarios tienen la obligación de:
1) Notificar toda sospecha de reacción adversa de la que tengan conocimiento durante su práctica habitual y enviarla lo más rápidamente posible al órgano competente en materia de farmacovigilancia de la Comunidad Autónoma correspondiente, mediante el formulario de recogida de sospechas de reacciones adversas ("tarjeta amarilla". Véase más adelante).
2) Conservar la documentación clínica de las sospechas de reacciones adversas a medicamentos, con el fin de completar o realizar el seguimiento, en caso necesario.
3) Cooperar con los técnicos del Sistema Español de Farmacovigilancia de medicamentos de uso humano, proporcionando la información necesaria que éstos les soliciten para ampliar o completar la información sobre la sospecha de reacción adversa.
4) Mantenerse informados sobre los datos de seguridad relativos a los medicamentos que habitualmente prescriban, dispensen o administren.
5) Colaborar con los responsables de farmacovigilancia de los titulares de autorizaciones de comercialización, en caso de una sospecha de reacción adversa a una de sus especialidades farmacéuticas, aportando la información que se precise para su posterior notificación al Sistema Español de Farmacovigilancia.
6) Colaborar, en calidad de expertos, con la Agencia Española del Medicamento y los órganos competentes de las Comunidades Autónomas, en la evaluación de los problemas de seguridad de los medicamentos de uso humano.
ALGUNAS DEFINICIONES ÚTILES EN FARMACOVIGILANCIA
Acontecimiento adverso
Experiencia indeseable que ocurre tras la administración de un medicamento. Un acontecimiento adverso no tiene que conllevar necesariamente una relación causal con el tratamiento8.
Información mínima de una reacción adversa
La notificación de una reacción adversa a un medicamento requiere, como mínimo, la información siguiente8:
Un profesional sanitario identificable como notificador. El notificador puede ser identificado por su nombre o iniciales, su dirección o su cualificación (por ejemplo, médico, dentista, farmacéutico, profesional de enfermería).
Un paciente identificable. Puede ser identificado por sus iniciales o por un número de paciente o por la fecha de nacimiento (o la edad si la fecha de nacimiento no se conoce) o por el sexo.
Al menos una sustancia o medicamento sospechoso.
Al menos una sospecha de reacción adversa.
Reacción adversa (fármaco comercializado)
Respuesta a un fármaco, que es nociva y no intencionada, y que se produce con las dosis utilizadas normalmente en el ser humano para la profilaxis, el diagnóstico o el tratamiento de enfermedades o para modificar una función fisiológica9. Las reacciones adversas a medicamentos se clasifican en seis tipos, que se identifican con las letras de la A a la F4,10:
Las reacciones de tipo A
Del inglés augmented. Son comunes y se hallan relacionadas con la acción farmacológica. Son predecibles y la mortalidad es usualmente (pero no invariablemente) baja. Ejemplos de reacciones adversas de tipo A son los efectos anticolinérgicos de los antidepresivos tricíclicos10, la bradicardia por bloqueadores beta y la hemorragia por anticoagulantes11.
Las reacciones de tipo B
Del inglés bizarre. Son poco frecuentes y pueden deberse a causas inmunológicas y farmacogenéticas, no se hallan relacionadas con la acción farmacológica ni con la dosis (aunque, en el segundo caso, las de causa genética pueden estarlo) y no son predecibles. La mortalidad puede ser elevada. Ejemplos de este tipo de reacciones son la hipersensibilidad a la penicilina, el rash cutáneo por ampicilina10, la hemólisis inducida por diversos fármacos, entre ellos la aspirina, en pacientes con deficiencia de glucosa-6-fosfato deshidrogenasa11 y la miocardiopatía por tacrolimus12.
Las reacciones de tipo C
Del inglés chronic. Son poco frecuentes y se hallan relacionadas con la dosis acumulativa del fármaco. Un ejemplo es la supresión del eje hipotálamo-hipófisis-suprarrenal por corticoides10.
Las reacciones de tipo D
Del inglés delayed. Son poco frecuentes, relacionadas con la dosis y ocurren o aparecen algún tiempo después de la utilización del fármaco. Ejemplos de este tipo de reacciones son el adenocarcinoma vaginal por dietilbestrol y la discinesia tardía por neurolépticos10.
Las reacciones de tipo E
Del inglés ending of use. Son poco frecuentes y ocurren tempranamente después de la supresión del fármaco. Ejemplos ilustrativos son el síndrome de abstinencia a opiáceos y la isquemia miocárdica por supresión de bloqueadores beta10.
Las reacciones de tipo F
Del inglés failure. Consisten en fracasos inesperados del tratamiento. Pueden producirse, entre otras causas, por uso inapropiado, interacciones o trastornos metabólicos13. Un ejemplo lo constituiría un embarazo no deseado debido a que la dosis de un contraceptivo oral podría haber sido insuficiente si se hubiera administrado concomitantemente un inductor enzimático10.
Reacción adversa grave
Reacción adversa que cumple uno de los siguientes criterios: ocasiona la muerte, pone en peligro la vida, requiere la hospitalización del paciente o prolonga la hospitalización ya existente, da lugar a una incapacidad o discapacidad persistente o significativa o constituye una anomalía congénita o defecto de nacimiento. También se consideran reacciones adversas graves aquellas que, sin cumplir ninguno de los criterios mencionados, se consideran importantes desde el punto de vista médico14.
Reacción adversa inesperada
Reacción adversa cuya naturaleza, gravedad o desenlace no sean coherentes con la información descrita en la ficha técnica8.
Señal
Información comunicada de una posible relación causal entre un acontecimiento adverso y un fármaco, cuando previamente esta relación era desconocida o había sido documentada de forma incompleta. Usualmente se requiere más de una notificación para generar una señal, dependiendo de la gravedad del acontecimiento y de la calidad de la información15.
Tarjeta amarilla
Formulario para la notificación de sospechas de reacciones adversas distribuida por los órganos competentes en materia de farmacovigilancia de las Comunidades Autónomas a los profesionales sanitarios7.
Evaluación de la relación de causalidad
Existen diversos argumentos por los cuales un profesional sanitario puede sospechar una relación causal entre una reacción adversa y un fármaco: la existencia de una secuencia temporal razonable entre la administración del fármaco y la aparición de una posible reacción adversa, la relación entre la discontinuación del fármaco y la desaparición de la reacción adversa (dechallenge), la recurrencia de la posible reacción adversa con la reintroducción del fármaco (rechallenge), la existencia de una relación entre la dosis y la gravedad de la posible reacción adversa, la existencia de una base farmacológica o toxicológica para la posible reacción adversa, el conocimiento de que un fármaco similar puede dar lugar a la reacción adversa sospechada (efecto de clase) y, por último, la ausencia de alternativas razonables que puedan explicar el acontecimiento, por ejemplo, tratamientos concomitantes, la enfermedad para la cual es administrado el fármaco sospechoso u otras enfermedades16. Tomando como base estos parámetros se han elaborado métodos para evaluar la relación de causalidad en casos individuales de sospechas de reacciones adversas. Entre los más conocidos se hallan el de Karch y Lasagna17, que clasifica la relación de causalidad como definitiva, probable, posible, condicional o no relacionada; el de Naranjo18, que la clasifica como definitiva, probable, posible o dudosa y el de la Organización Mundial de la Salud (OMS)19, cuyas categorías de clasificación son cierta, probable, posible, poco probable, condicional/no clasificada, no valorable/inclasificable. De todas formas, en un paciente determinado es con frecuencia imposible demostrar que el fármaco es el agente causal4.
Detección de señales
Las sospechas de reacciones adversas son notificadas por los profesionales sanitarios a los órganos competentes en materia de farmacovigilancia de la Comunidades Autónomas, donde las notificaciones son codificadas, evaluadas y remitidas a la Agencia Española del Medicamento que, a su vez, las envía al Centro Internacional de la OMS en Uppsala, cuyo objetivo principal es la detección precoz de las reacciones adversas nuevas de los nuevos medicamentos y vigilar las reacciones adversas graves y raras de todos los medicamentos, alertando a las autoridades sanitarias para que tomen medidas que las eviten. Las principales limitaciones de la notificación espontánea son la infranotificación y la dificultad para evaluar la relación de causalidad4.
El objetivo principal de la notificación espontánea de reacciones adversas es la detección precoz de las señales de problemas relacionados con la seguridad de los medicamentos que no han podido ser identificadas durante la etapa de desarrollo clínico previa a la comercialización. En la actualidad existen numerosas bases de datos que recogen sospechas de reacciones adversas en un número que se sabe que es inferior a las que realmente se producen debido a la infranotificación, pero que en conjunto suponen una gran información20. La base de datos FEDRA (Farmacovigilancia Española Datos de Reacciones Adversas) es la del Sistema Español de Farmacovigilancia.
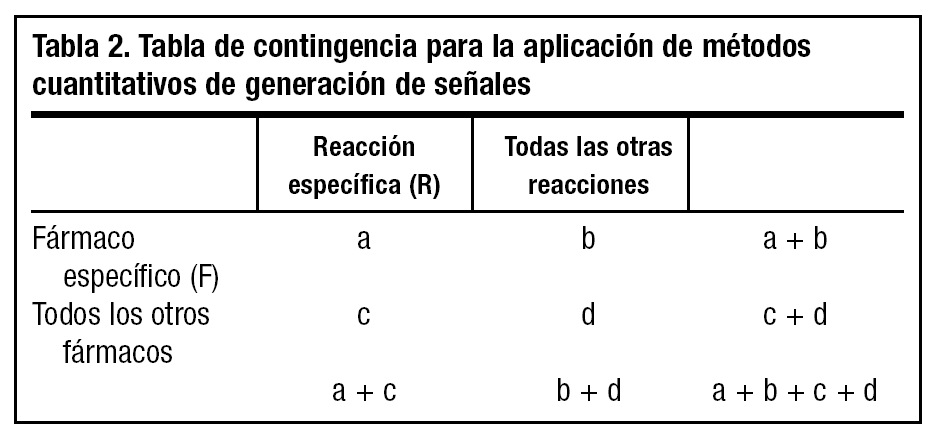
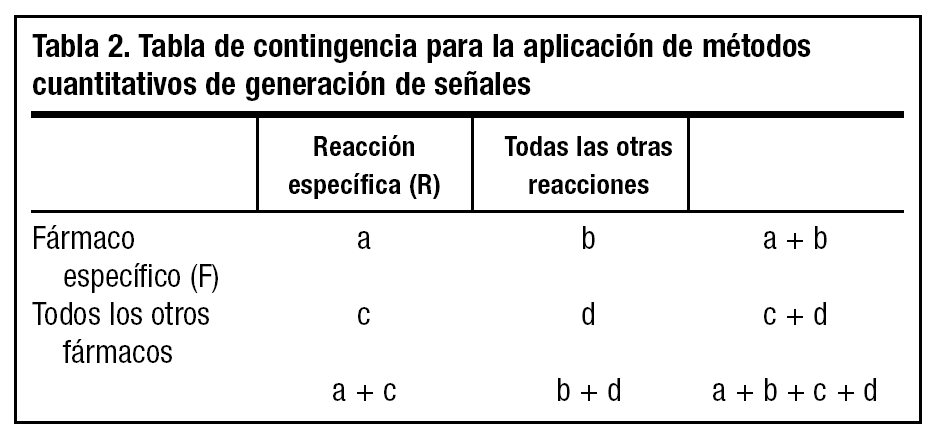
Se han propuesto diferentes métodos cuantitativos de detección de señales basados en la aplicación de procedimientos matemáticos que buscan en las bases de datos desproporcionalidades estadísticas que orientan a la existencia de una posible señal20,21. Estos procedimientos ofrecen unos estimadores que tienen como única función alertar y que, por supuesto, no implican necesariamente una relación causal. El cálculo de la medida de desproporcionalidad en todos estos procedimientos se basa en una tabla de contingencia de dos por dos, en la que en la celda "a" se disponen las notificaciones en las que están asociadas la reacción específica (R) y el fármaco específico (F), en la celda "b" las notificaciones de otras reacciones asociadas a F, en la celda "c" las de R asociada a otros fármacos y en la "d" las de otras reacciones asociadas a otros fármacos (tabla 2) . Así, pueden calcularse estimadores de desproporcionalidad tales como la relación entre las proporciones de notificación (proportional reporting ratio [PRR] = [a/{a + b}]/[c/{c+d}]), la odds ratio de notificación (reporting odds ratio [ROR] = [a x d]/[c x b]) y el componente de información (CI), que es el logaritmo de la relación entre la frecuencia de notificación observada de una asociación fármaco-reacción adversa y la frecuencia esperada en base al número de notificaciones de ese fármaco y de esa reacción, bajo la hipótesis nula de no asociación entre fármaco y reacción, en cuyo caso el CI es 020. Mediante cada uno de estos procedimientos se estima que se ha detectado una desproporcionalidad cuando PRR-1,96 error estándar (EE) > 1 o cuando ROR-1,96 EE > 1 o cuando CI-2 desviaciones estándar > 021. No debe olvidarse, no obstante, que estos procedimientos indican, únicamente, la probabilidad de que una asociación fármaco-reacción adversa sea una señal y merezca una evaluación cuidadosa y en profundidad mediante estudios específicos tales como estudios epidemiológicos de campo o en bases de datos, ensayos clínicos u otros tipos de estudios4.
Gestión del riesgo en farmacovigilancia
Una vez identificada y confirmada una señal existen tres acciones relevantes: a) adoptar las medidas administrativas de reducción del riesgo; b) comunicar a los profesionales sanitarios y a los pacientes la existencia del riesgo, las medidas adoptadas y las recomendaciones al respecto, y c) establecer las estrategias específicas para su prevención3. Dependiendo de si el riesgo es aceptable en las condiciones de uso autorizadas, de si es aceptable en ciertas situaciones o de si es inaceptable, se llevan a cabo medidas reguladoras que pueden consistir en la información sobre la reacción adversa y las medidas para prevenirla, si se conocen, la restricción de indicaciones, introducción de contraindicaciones, restricción a ciertos grupos de población, realización de pruebas clínicas o analíticas, restricción del ámbito de la prescripción (diagnóstico hospitalario, uso hospitalario, prescripción por especialista), restricción de ciertas presentaciones o la retirada del fármaco del mercado.
Consideraciones finales
Es indudable que el conocimiento sobre los beneficios y riesgos de los fármacos constituye un área del máximo interés para todas aquellas personas o entidades que desarrollan su labor profesional en el área de la salud, tales como los clínicos, la industria farmacéutica y las autoridades sanitarias y, en último término, para la sociedad. Por ello, es importante que los clínicos y, en general, los profesionales sanitarios notifiquen las sospechas de reacciones adversas que puedan detectar en la práctica. Sin duda, vale la pena invertir tiempo y esfuerzo para contribuir a la utilización racional de los medicamentos.
Correspondencia: J. Borja Villegas.
Departamento de Farmacovigilancia. Unidad de Investigación
Clínica. J. Uriach y Compañía, S.A.
Polígon Industrial Riera de Caldes. Avinguda Camí Reial, 51-57.
08184 Palau-solità i Plegamans. Barcelona.
Correo electrónico: fv-borja@uriach.com
Recibido el 19-10-05; aceptado para su publicación el 15-02-06.
