





INTRODUCCIÓN
La insuficiencia suprarrenal primaria (ISP) es una enfermedad bastante rara, siendo sus causas más frecuentes la autoinmunitaria y las infecciones como la tuberculosis. Más raramente todavía forma parte del síndrome pluriglandular autoinmune (SPA), donde la ISP se asocia a enfermedad tiroidea autoinmune en el 69% de los casos, y a otras enfermedades mediadas autoinmunológicamente como el hipoparatiroidismo, hipogonadismo, diabetes mellitus tipo 1, enfermedad de Graves, etc., teniendo que coexistir dos o más de estas enfermedades para llegar al diagnóstico1. Existen dos tipos de SPA, el tipo 1 que se presenta esporádicamente o de forma autosómica recesiva, y el tipo 2 que se hereda de forma autosómica dominante y se ha asociado a los HLA DR3 y DR42. Presentamos el caso clínico de una paciente que consulta por hiperpigmentación difusa como primera manifestación de una ISP en el contexto de un SPA, a partir del diagnóstico realizado en Atención Primaria.
EXPOSICION DEL CASO
Mujer de 20 años de edad y nacionalidad rumana, que desde hace un año y medio coincidiendo con su llegada a España, ha notado aumento de la pigmentación de la piel y de las mucosas en toda la superficie corporal. En una segunda consulta la paciente además refiere en los últimos meses astenia, estreñimiento, somnolencia y aumento de peso.
Entre sus antecedentes personales destacaba una intervención quirúrgica por hernia umbilical en la infancia. No presentaba alergias conocidas, ni tomaba ningún tipo de medicamento. Tampoco presentaba alteraciones menstruales ni otras enfermedades de interés. No existían antecedentes familiares importantes.
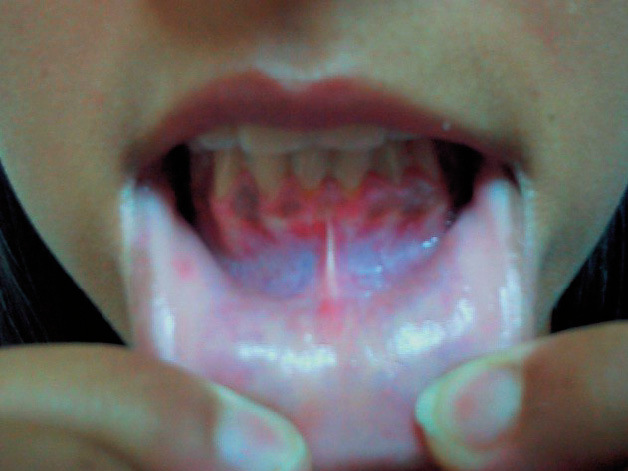
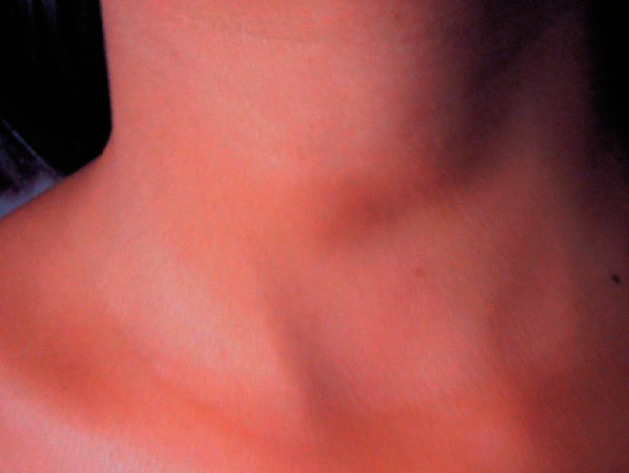
A la exploración se observó un aumento de la pigmentación de forma difusa en toda la superficie corporal, incluso en zonas no fotoexpuestas y en pliegues, así como pigmentación de encías (fig. 1). No presentaba candidiasis mucocutánea. A nivel cervical se apreció un aumento del tamaño del tiroides de forma difusa a la palpación, sin nódulos (fig. 2). La exploración física reveló también una tensión arterial (TA) de 90/60 mmHg, un peso de 52 kg, y una talla de 162 cm, siendo el resto de la exploración normal. Se solicitó un hemograma, y una bioquímica general con perfil hepático, iones, hierro y ferritina que resultaron ser normales, así como el sedimento de orina.
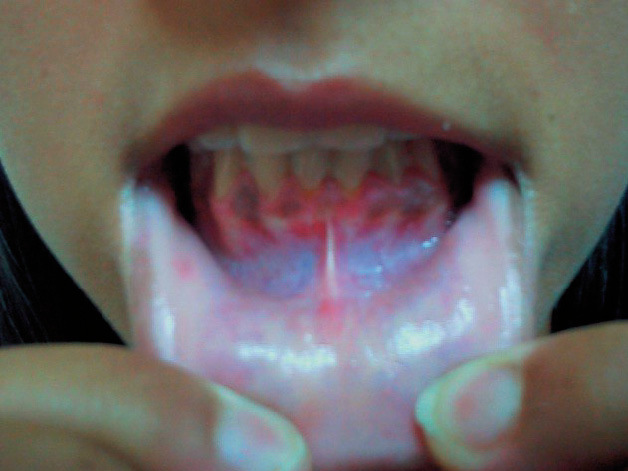
Figura 1. Hiperpigmentación de encías característica del Addison.
Figura 2. Aumento del tamaño del tiroides de forma difusa.
En el estudio hormonal se observaron los siguientes resultados: hormona estimuladora de tiroides (TSH) = 14,14 µU/ml (valor negativo -vn-: 0,49-4,67); T4 libre = 0,50 ng/dl (vn: 0,71-1,85); hormona adrenocirticotrópica (ACTH) > 1.500 pg/ml (vn: < 80); cortisol basal < 1 µg/dl (vn: 5-25); cortisol libre en orina de 24 horas = 1,2 µg/24h (vn: 20-100); hormona paratiroidea (PTH) = 8,76 pg/ml (vn: 14-60); siendo normales la hormona foliculoestimulante (FSH), hormona luteinizante (LH), prolactina (PRL) y estradiol. En el estudio inmunológico se observaron unos anticuerpos antiadrenales positivos 1/80; anticuerpos antinucleares positivos 1/160 con patrón homogéneo; anticuerpos antitiroglobulina de 427 UI/ml (vn:0-400), y anticuerpos antimicrosomales de 236 UI/ml (vn:0-100). Se realizó un Mantoux que fue positivo con radiografía de tórax normal.
A la vista de todos los resultados se diagnosticó de insuficiencia suprarrenal primaria autoinmune, hipotiroidismo e hipoparatiroidismo primarios autoinmunes, en el contexto de un SPA. El SPA parece ser del tipo 2 ya que, a diferencia del tipo 1, es más frecuente en mujeres, aparece en la edad adulta, y no presenta la candidiasis mucocutánea característica del tipo 1. La determinación de los HLA DR3 y DR4 confirmarán el diagnóstico.
DISCUSIÓN
La ISP se debe a una secreción de hormonas corticosuprarrenales inferior a las demandas del organismo. Es una enfermedad que se manifiesta clínicamente con hipotensión, hiperpigmentación, astenia, pérdida de peso y anorexia; evoluciona de forma progresiva con episodios de reagudización (crisis addisoniana) ante situaciones de estrés, y sin un tratamiento adecuado puede conducir a la muerte.
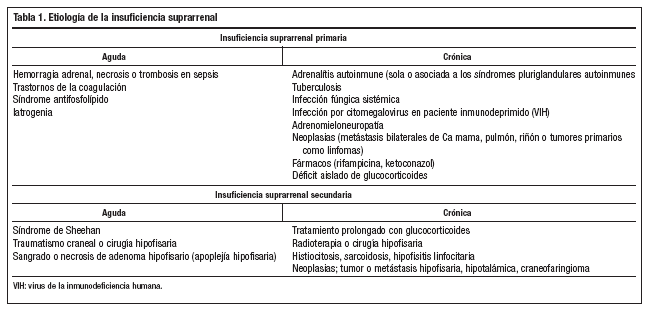
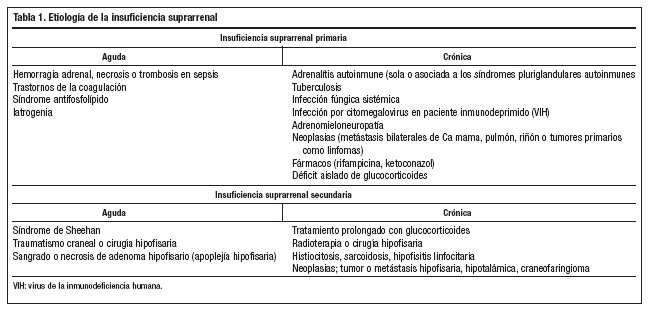
Tiene una incidencia de 0,83 casos/100.000 habitantes y una prevalencia de 4-6 casos/100.000 habitantes3; la edad media de aparición es 40 años aunque puede aparecer a cualquier edad. Actualmente la etiología más frecuente es la adrenalitis autoinmune, seguida por la tuberculosa, y posteriormente otras causas como infección fúngica sistémica, virus de la inmunodeficiencia humana (VIH), neoplasias y fármacos (tabla 1). En la adrenalitis autoinmune se encuentran anticuerpos contra la glándula suprarrenal hasta en un 65% de los casos con carácter diagnóstico. La adrenalitis autoinmune en más del 50% de los casos se asocia a otros déficit endocrinos, formando parte de los SPA tipos 1 y 2.
El SPA tipo 1 se caracteriza por hipoparatiroidismo, candidiasis mucocutánea e insuficiencia suprarrenal como forma de presentación más frecuente, mientras que el SPA tipo 2 consiste en la asociación de insuficiencia adrenal y tiroiditis autoinmune, asociado en ocasiones a DM tipo 1 y/o hipogonadismo (tabla 2)2.

Las manifestaciones clínicas suelen cursar de forma insidiosa durante años. Los pacientes consultan principalmente por astenia, hiperpigmentación, anorexia, fatiga, trastornos gastrointestinales, hipotensión y humor depresivo que, cuando aparecen de forma conjunta nos debe hacer pensar en una insuficiencia suprarrenal4. Los síntomas pueden agruparse según el déficit que los produce:
1) Por déficit de glucocorticoides, con la hiperpigmentación de piel y mucosas característica del Addison (en zonas expuestas, pliegues palmares, nudillos, alrededor de las uñas, mucosa genital, areolas mamarias, mucosa bucal y en lengua, y también en cicatrices producidas durante el desarrollo de la enfermedad)5. Dicha hiperpigmentación está producida por un aumento del precursor del ACTH, que produce ACTH y ß-lipotropina (contiene hormona que estimula los melanocitos [MSH]). El déficit de cortisol produce hipoglucemia, astenia y fatiga muscular. También aparecen síntomas digestivos como dolor abdominal, náuseas, diarrea y/o estreñimiento; y síntomas psíquicos como fatiga mental, insomnio, irritabilidad o humor depresivo.
2) Por defecto mineralcorticoide, con hipotensión arterial, hipotensión ortostática, taquicardia, palpitaciones o síncope6.
3) Por déficit de andrógenos, con pérdida del vello axilar y pubiano, caída de cabello, oligomenorrea e inhibición de la libido.
4) Otros síntomas, en función de la etiología (tumores, sepsis...) y/o los defectos endocrinos que acompañen a la insuficiencia suprarrenal primaria autoinmune.
En los datos de laboratorio se aprecia anemia normocítica y normocrómica; además de eosinofilia y velocidad de sedimentación globular (VSG) aumentada, hipoglucemia, hiponatremia, hiperpotasemia, e hipercalcemia. En el SPA tipo 2 puede existir anemia macrocítica por la presencia de anticuerpos frente a las células parietales, y déficit de absorción de vitamina B12.
Tras la sospecha clínica y el análisis bioquímico un cortisol plasmático basal < 3 µg/dl y un ACTH basal > 100 pg/ml nos dará el diagnóstico; mientras que un cortisol plasmático basal > 19 µg/dl nos excluirá el mismo. Sin embargo un ACTH basal normal no excluye el diagnóstico1.
Si con estas determinaciones no llegamos al diagnóstico (cortisol entre 3 y 19 µg/dl, o en insuficiencia suprarrenal de reciente comienzo), se realizarán pruebas de estimulación hormonal 7 como:
1) Prueba de estimulación rápida con ACTH: se administran 250 µg por vía intramuscular o intravenosa de ACTH sintético y se mide el cortisol plasmático basal, y a los 30 y 60 minutos. Si el cortisol plasmático es > 19 µg/dl en cualquier extracción se considera normal.
2) Prueba de hipoglucemia insulínica: se administra 0,1-0,15 UI/kg de peso de insulina rápida, provocando una hipoglucemia clínica y bioquímica. Se mide cortisol plasmático basal, y a los 30, 60 y 90 minutos. Se considera normal si el cortisol plasmático es > 18-20 µg/dl en cualquier extracción.
Una vez confirmada la insuficiencia suprarrenal, investigaremos la causa que la está provocando. Para ello podemos realizar los siguientes estudios:
1) Anticuerpos antisuprarrenales: tienen una alta especificidad y sensibilidad teniendo carácter diagnóstico de adrenalitis autoinmune; sin embargo disminuyen a medida que evoluciona la enfermedad, por lo que en fases avanzadas no son representativos y nos guiaremos por la clínica y el resto de exploraciones.
Actualmente se estudian anticuerpos frente a enzimas esteroidogénicas8, tales como 17 *-hidroxilasa, 21 hidroxilasa9 y citocromo P450 con escisión de la cadena lateral, ya que parecen ser más específicos.
El Addison autoinmune aumenta el riesgo de padecer otras enfermedades autoinmunes, por lo que sería conveniente solicitar anticuerpos frente a otros órganos en el seguimiento de estos pacientes.
2) Tomografía axial computarizada (TAC): se debe solicitar en el diagnóstico etiológico del Addison, excepto en el caso de adrenalitis autoinmune o adrenomieloneuropatía. Existen tres patrones radiológicos que nos ayudan a identificar la etiología 10: a) Atrofia suprarrenal: debemos sospechar una adrenalitis autoinmune, aunque puede ser una secuela de hemorragias suprarrenales, de tuberculosis o de otras enfermedades granulomatosas. b) Calcificación: la más frecuente es la de tipo tuberculosa y en menor medida como secuela de hemorragias, infecciones micóticas
en inmunodeprimidos y algunos procesos neoplásicos.
c) Agrandamiento suprarrenal: tuberculosis de pocos años de evolución, metástasis o neoplasias primarias, linfomas, hemorragias suprarrenales recientes, infecciones micóticas y víricas, amiloidosis, sarcoidosis.
3) Otras pruebas según la etiología: biopsia con punción-aspiración con aguja fina (PAAF), resonancia magnética nuclear (RMN), prueba de la tuberculina, radiografía de tórax, hormonas tiroideas, metabolismo hidrocarbonado, fosfocálcico, etc.
El tratamiento de la crisis suprarrenal va a consistir en la administración de 100 mg de hidrocortisona en bolo intravenoso, para posteriormente añadir 200-300 mg en el suero fisiológico en las siguientes 24 horas. Por encima de 60 mg/día de hidrocortisona no es necesario administrar fluorhidrocortisona. Además hay que realizar una reposición electrolítica, y tratar la causa desencadenante.
El tratamiento de mantenimiento, consistirá en la administración de 20-30 mg/día de hidrocortisona (repartido en dos tercios por la mañana y un tercio por la tarde), para posteriormente ir disminuyendo hasta conseguir la menor dosis que mantenga sin síntomas al paciente. También se puede utilizar cortisona a una dosis de 25-35 mg/día. Se debe aumentar la dosis corticoidea ante situaciones de estrés como infecciones, cirugía, partos, etc. Además se debe instaurar tratamiento con fluorhidrocortisona 50-200 µg/día en una sola toma diaria. La dosis se mantendrá según la TA, la actividad de la renina plasmática y el potasio.
Por último, todos los pacientes deberían llevar una tarjeta o placa identificativa donde se especifiquen las recomendaciones sobre cómo actuar ante posibles complicaciones.
Resumiendo:
1) La ISP es una enfermedad bastante rara, siendo sus causas más frecuentes la autoinmunitaria y las infecciones como la tuberculosis.
2) En todo paciente diagnosticado de ISP cuya causa sea autoinmunitaria, hay que descartar la existencia de otros déficits endocrinos de causa autoinmune, ya que en más del 50% de los casos la ISP se asocia a otras patologías de origen autoinmune como la tiroiditis crónica, el hipoparatiroidismo autoinmune y la diabetes tipo 1, formando parte de los SPA tipos 1 y 2.
3) Destacar la importancia de la historia clínica y la sospecha de ISP ante una paciente con hiperpigmentación de piel y mucosas generalizada, gracias a lo cual el médico de Atención Primaria tuvo una visión global de la biografía de la paciente, pudiendo llegar de ese modo al diagnóstico.
4) Creemos que es importante crear vías de comunicación entre los médicos de Atención Primaria y Especializada para conseguir un adecuado nivel de calidad en la atención al paciente.
