








tracción; el tipo de muestra utilizada y las necesidades de precisión analítica. También se señalan algunos aspectos relativos a la farmacocinética clínica de los antiepilépticos (absorción, distribución y eliminación), factores que modifican la farmacocinética y la relación dosis-nivel sérico, tipos de cinética, índices nivel sérico/dosis y rangos terapéuticos.
CONCEPTO Y ANTECEDENTES
La monitorización terapéutica de niveles séricos (therapeutic drug monitoring [TDM]) de los fármacos antiepilépticos consiste en la determinación periódica de la concentración de los antiepilépticos en suero, con la finalidad de mejorar la eficacia y seguridad del tratamiento en cada paciente.
La TDM de antiepilépticos comenzó hace unos 30 años y constituye un procedimiento clínico habitual, que ha contribuido a mejorar la utilización racional de estos fármacos1. Las técnicas analíticas empleadas al principio fueron las cromatográficas y después las inmunoanalíticas.
En los años 80, el desarrollo tecnológico aumentó la efectividad de la TDM, mediante la disponibilidad de autoanalizadores y microordenadores. Este hecho permitió difundir la TDM a numerosos laboratorios, mejorar la exactitud y precisión de las determinaciones, disminuir los tiempos de respuesta, facilitar el mantenimiento de bases de datos y agilizar los cálculos farmacocinéticos2.
FUNDAMENTO
La TDM de antiepilépticos se fundamenta en las siguientes razones3,4:
La valoración de los efectos farmacológicos por criterios clínicos es difícil y lenta, sobre todo si las crisis son infrecuentes, en la prevención de crisis febriles o convulsiones agudas tras traumatismo craneoencefálico o neurocirugía. Además, unas crisis epilépticas frecuentes pueden deberse tanto a ineficacia como a toxicidad del tratamiento5,6.
Existe una mejor relación del efecto farmacológico con el nivel sérico del antiepiléptico que con la dosis prescrita, por las diferencias farmacocinéticas interindividuales y por la cinética no lineal de algunos compuestos.
Los anticonvulsivantes más utilizados presentan un margen terapéutico estrecho: las concentraciones mínima eficaz y mínima tóxica están próximas.
Es difícil la valoración clínica de la toxicidad en neonatos y pacientes con alteraciones neurológicas, demencia o retraso psicomotor. A veces, la toxicidad ha sido atribuida incorrectamente a la enfermedad.
Es necesario prevenir las reacciones tóxicas graves, como las lesiones cerebelosas irreversibles causadas por la fenitoína.
Para varios antiepilépticos, su efecto es proporcional a la concentración en el sitio de acción: el sistema nervioso central.
Los antiepilépticos tradicionales tienen un rango terapéutico conocido, mejor llamado intervalo óptimo, que incluye las concentraciones séricas en las que, en la mayoría de individuos, hay eficacia (buen control de las crisis) y ausencia de toxicidad. No obstante, el intervalo óptimo es distinto al intervalo de valores normales de un parámetro bioquímico. Sus límites no pueden aplicarse con rigidez de forma individualizada, pues hay una superposición entre los rangos teóricamente asociados con eficacia y los niveles supra e infraterapéuticos. Así, un paciente con concentraciones séricas del antiepiléptico situadas bajo el límite inferior del rango terapéutico puede tener sus crisis bien controladas y otro con concentraciones situadas dentro del rango terapéutico puede presentar toxicidad.
Muchos rangos terapéuticos publicados han sido obtenidos en pacientes con epilepsia refractaria, mientras que los pacientes con epilepsia menos grave pueden responder a concentraciones séricas menores, situadas por debajo del llamado límite terapéutico inferior. Por ello, hay dos tendencias actuales que impulsan a una interpretación flexible de los rangos terapéuticos (tabla 1). Estas tendencias consisten en: a) considerar potencialmente terapéutica cualquier concentración que no alcance el límite terapéutico superior, "techo" que podría incluso superarse en situaciones clínicas concretas7 y b) establecer dos intervalos o rangos concéntricos: el interno sería el intervalo óptimo (rango de concentraciones deseables para una mayoría de pacientes) y el externo incluiría también unos límites de concentraciones extremas que podrían alcanzarse eventualmente y ser eficaces sin producir toxicidad concentraciones altas) o sin alcanzar el límite terapéutico inferior (concentraciones bajas)8.

ASPECTOS CLINICOS DE LA FARMACOCINÉTICA
Procesos que comprende
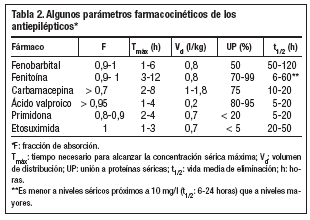
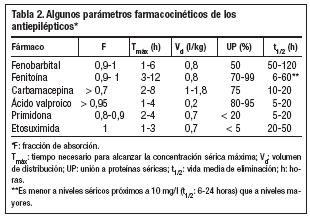
Los antiepilépticos suelen administrarse por vía oral. Después de su administración se liberan de su forma farmacéutica, se solubilizan en los fluidos biológicos y sufren una serie de procesos (absorción, distribución, metabolización y excreción), que determinan en cada momento su concentración en suero y otros líquidos del organismo. Cada fármaco, también los antiepilépticos, presenta unos parámetros farmacocinéticos que lo individualizan9-12. Algunos de estos parámetros figuran en la tabla 2.
Absorción
La absorción gastrointestinal de los antiepilépticos suele ser buena. Su fracción de absorción (F) es la proporción entre la cantidad de fármaco absorbida por vía oral respecto a la cantidad absorbida por vía intravenosa. Es un índice de la cantidad absorbida y suele ser alta, próxima a 1, con la excepción de la carbamacepina, cuya absorción es irregular (F = 0,7 en algunos casos).
La velocidad de absorción de los antiepilépticos es variable, rápida para el ácido valproico y etosuximida y en el extremo opuesto, la fenitoína y carbamacepina.
El tiempo necesario para alcanzar la concentración sérica máxima tras una dosis (Tmáx) depende de las velocidades de absorción y eliminación, pues dicha concentración se alcanza cuando se igualan ambas velocidades.
Algunos factores que pueden modificar la absorción oral de los antiepilépticos son:
La presencia de alimentos en el estómago suele disminuirla (valproato). Sin embargo, para la fenitoína o carbamacepina, antiepilépticos poco hidrosolubles, puede aumentar su disolución y posterior absorción.
La nutrición enteral administrada por sonda nasogástrica puede disminuirla (fenitoína).
El preparado farmacéutico utilizado. Pueden presentarse diferencias en la biodisponibilidad para los distintos preparados comerciales de fenitoína, carbamacepina y fenobarbital. El cambio de excipiente en un preparado de fenitoína causó un aumento considerable de la incidencia de reacciones tóxicas debidas a este antiepiléptico, por haber aumentado su biodisponibilidad. También es previsible la absorción más rápida del antiepiléptico al emplear preparados magistrales, lo que es relativamente frecuente en nuestro país para la fenitoína. La utilización de comprimidos de valproico con cubierta entérica retrasa la absorción de 1-2 horas (comprimidos sin cubierta entérica) a 4-8 horas.
La dosis utilizada. La biodisponibilidad de carbamacepina y fenitoína tras su administración oral disminuye al aumentar la dosis.
Distribución
Una vez absorbidos, los antiepilépticos pasan desde el suero a los distintos órganos y tejidos. En el suero se unen a las proteínas, principalmente a la albúmina, y en proporción menor a la *1-glucoproteína ácida.
El volumen de distribución aparente refleja la cantidad de fármaco presente en los tejidos en relación a la concentración sérica. Dicho volumen depende: directamente del volumen de líquido corporal en el que se distribuye realmente y del grado de unión del fármaco a los tejidos, e inversamente de su unión a las proteínas séricas. El valproato es el antiepiléptico que tiene un volumen de distribución menor (0,2 l/kg). Los principales factores que influyen en el volumen de distribución son la edad (en los ancianos disminuye el agua corporal y aumenta el tejido graso), el embarazo (en el tercer trimestre aumenta el agua corporal y disminuye la unión a proteínas por la hipoalbuminemia) y la unión a proteínas.
Sólo el fármaco libre (no unido a las proteínas séricas) penetra en los tejidos y llega al sitio de acción. Los antiepilépticos que se unen a las proteínas séricas en mayor proporción son fenitoína y ácido valproico. El porcentaje de unión de estos dos últimos varía notablemente según los individuos (fenitoína) o la concentración alcanzada (ácido valproico). Los porcentajes de unión más bajos corresponden a la primidona (< 20%) y la etosuximida (no se une a las proteínas séricas).
La unión a proteínas séricas de varios antiepilépticos (fenitoína, valproico) disminuye por diversos factores, que pueden aumentar su fracción libre y su eficacia, produciendo a veces toxicidad. Estos factores pueden ser: fisiológicos (embarazo, neonatos, ancianos), patológicos (hipoalbuminemia, hiperbilirrubinemia, nefropatía, cirrosis) o farmacológicos (interacción entre valproico y fenitoína).
Eliminación
La vía de eliminación principal para los antiepilépticos es la biotransformación por los microsomas hepáticos. La excreción urinaria del fármaco sin biotransformar sólo tiene importancia para la primidona y el fenobarbital.
La metabolización de carbamacepina y primidona produce metabolitos activos (carbamacepina-10,11-epóxido para la carbamacepina; fenobarbital y feniletilmalonamida para el fenobarbital) cuya proporción depende de la variabilidad interindividual en la capacidad de biotransformación y del tratamiento asociado con otros fármacos. Estos metabolitos activos influyen en la eficacia del tratamiento y en la posible aparición de efectos tóxicos.
El parámetro más usual para estimar la eliminación de un fármaco es la vida media, que se define como el tiempo necesario para que la concentración sérica del fármaco disminuya a la mitad.
Tipos de cinética10
Lineal
La intensidad y la velocidad de los procesos farmacocinéticos (absorción, distribución, eliminación) no varía con el tiempo ni con la dosis. Para los fármacos que la siguen, hay una relación lineal, directamente proporcional, entre la dosis administrada y el nivel sérico estable alcanzado.
No lineal
Las constantes farmacocinéticas (vida media, unión a proteínas, etc.) varían con el tiempo o con la dosis.
Dependiente de la dosis: puede ser creciente o decreciente, según que el nivel sérico del fármaco aumente, respectivamente, más o menos de lo previsible por el aumento realizado en la dosis prescrita.
Un ejemplo de la primera es la biotransformación de la fenitoína, que se satura y disminuye desproporcionadamente por encima de una concentración sérica cercana a su límite terapéutico inferior (8-10 mg/l). Este hecho implica un riesgo de toxicidad, al aumentar los niveles de fenitoína más de lo previsible para un discreto aumento de dosis prescrita.
Un ejemplo de la decreciente es la saturación de la unión a proteínas del ácido valproico, cuya fracción libre aumenta transitoriamente cuando su concentración sérica se incrementa.
Dependiente del tiempo: el caso de la carbamacepina es el más representativo. Su eliminación aumenta progresivamente durante el primer mes de tratamiento, por autoinducción enzimática de su metabolización. Esto tiene dos consecuencias: una vida media más larga tras la administración de una sola dosis de carbamacepina (24-50 horas) que en el tratamiento crónico (10-20 horas) y la obtención de niveles séricos menores en tratamientos que se prolonguen más de un mes. Por esta razón se recomienda comenzar el tratamiento con carbamacepina con dosis bajas y aumentarlas gradualmente para alcanzar la dosis deseada al mismo tiempo que la autoinducción máxima y evitar así fluctuaciones en la eficacia conseguida.
UTILIDAD DE LA RELACION DOSIS-NIVEL SÉRICO
La gran variabilidad farmacocinética interindividual dificulta la predicción de la concentración sérica que puede obtenerse con una dosis determinada en cada paciente. Los antiepilépticos que tienen una relación dosis-nivel sérico más pobre son la fenitoína y el valproato, por los siguientes motivos: su cinética dosis dependiente dificulta la predicción de los niveles alcanzados, al no variar éstos en proporción a los cambios de la dosis administrada; sufren interacciones que pueden afectar simultáneamente a varios procesos farmacocinéticos (unión a proteínas y biotransformación); tienen un alto grado de unión a las proteínas séricas, que aumenta la posibilidad de un desplazamiento de su unión a proteínas con el consecuente aumento de sus concentraciones libres.
Factores que determinan la relación dosis-nivel sérico
Diferencias farmacocinéticas individuales9,13
Genéticas. Se conocen familias con varios miembros que presentaron fenómenos tóxicos por incapacidad de hidroxilación de la fenitoína. Su enzima metabolizadora (fenitoína hidroxilasa) puede presentar dos grados de actividad enzimática que originan dos poblaciones (metabolizadores lentos y rápidos).
Edad. El metabolismo hepático evoluciona con la edad. Así, es bajo en el recién nacido y aumenta progresivamente durante los primeros meses. En los recién nacidos, sobre todo en los prematuros, y en el niño pequeño deben emplearse menores dosis de antiepilépticos que las correspondientes a su peso; en ellos, además de una menor biotransformación, hay también una hipoalbuminemia que aumenta la fracción libre y los efectos del fármaco. En los niños mayores de un año el metabolismo de los fármacos es mayor, necesitando emplear dosis más altas en los niños de 1 a 9 años que en los adultos. La capacidad metabólica disminuye progresivamente desde los 3 o 4 años hasta la adolescencia, en que se hace semejante a los adultos.
Aunque se reconoce una capacidad de biotransformación menor en los ancianos, que alarga la vida media de muchos fármacos, como fenitoína y valproato, no suele tener trascendencia práctica porque también hay una reducción de su unión a las proteínas séricas.
Menstruación. Disminuyen los niveles séricos de fenitoína.
Embarazo. En él se modifican 3 parámetros farmacocinéticos: disminuye la albúmina sérica, aumenta la capacidad metabólica hepática en el último trimestre y aumenta el líquido corporal. También aumenta el peso de la paciente y suele aceptarse que la concentración sérica total de los antiepilépticos disminuye a partir del sexto mes de gestación.
Parto. Tras él disminuye progresivamente en la madre la biotransformación de los antiepilépticos, normalizándose en unas semanas.
Fiebre. Aumenta la biotransformación y disminuyen los niveles séricos de fenitoína, carbamacepina y fenobarbital.
Enfermedad hepática. La hepatopatía crónica y/o grave puede modificar los niveles séricos de antiepilépticos de 3 maneras: disminuyendo la capacidad metabólica del hepatocito, que requiere bajar la dosis del antiepiléptico; disminuyendo la unión a las proteínas séricas (por hipoalbuminemia o por la competencia de sustancias endógenas como la bilirrubina), que aumenta la concentración de fármaco libre; aumentando el volumen de distribución, debido a la ascitis y/o al edema.
La cirrosis aumenta la vida media de fenobarbital y ácido valproico, y disminuye la unión a proteínas de este último. La hepatitis vírica aguda disminuye el metabolismo de ácido valproico y primidona.
Enfermedad renal. Pueden producirse 3 alteraciones farmacocinéticas: disminución de la excreción renal, que tiene importancia para la primidona y su metabolito feniletilmalonamida; disminución de la unión a proteínas séricas por hipoalbuminemia, por la competencia de sustancias endógenas (péptidos y ácidos grasos) o por alteraciones estructurales de la albúmina (carbamilación); aumento del metabolismo hepático.
En pacientes con insuficiencia renal, la fenitoína presenta un aclaramiento y un volumen de distribución mayores, una vida media más corta y una disminución de su unión a las proteínas séricas.
La disminución de la unión a proteínas séricas de los antiepilépticos de carácter ácido no mejora con la diálisis y sólo lo hace parcialmente con el trasplante renal. Por otro lado, en los pacientes con insuficiencia renal se ha demostrado el aumento de la fracción libre de carbamacepina y ácido valproico, así como la disminución de los niveles estables de carbamacepina y fenitoína.
Interacciones farmacológicas
Los niveles séricos de los antiepilépticos pueden cambiar significativamente por la administración simultánea de otros fármacos, que pueden dar lugar a interacciones farmacocinéticas14,15. Los mecanismos causales más destacables de las mismas son:
Inducción enzimática por los propios fármacos antiepilépticos (fenobarbital, fenitoína, carbamacepina, primidona) y por el etanol. El resultado suele ser la disminución del nivel sérico del antiepiléptico que sufre la inducción, que obliga a emplear dosis mayores para mantener los niveles séricos y la eficacia similares a los valores previos a la administración simultánea del fármaco inductor.
Inhibición enzimática del metabolismo de los antiepilépticos por fármacos como la isoniacida, eritromicina y ácido valproico. La consecuencia es el aumento de los niveles séricos de los antiepilépticos, que conduce a emplear dosis menores de los fármacos inhibidores. Este aumento no siempre ocurre, porque también pueden interactuar otros mecanismos farmacocinéticos que modifican el efecto final. Por ejemplo, el valproato y la fenilbutazona, que disminuyen la unión a proteínas de la fenitoína y, por tanto, sus niveles totales. En la interacción valproato-fenitoína suele predominar el efecto inhibidor de la metabolización de la fenitoína ejercido por el valproato antes que el efecto desplazante de la unión a la albúmina.
Desplazamiento de la unión a proteínas séricas de antiepilépticos (fenitoína y valproato), por parte de otros fármacos (warfarina, cloracepato, dicumarol, furosemida) que se unan al mismo sitio de fijación de la albúmina que los antiepilépticos citados. Este desplazamiento puede producir un aumento transitorio de la concentración libre y la eficacia del antiepiléptico, que se compensa con una mayor eliminación metabólica del antiepiléptico desplazado, produciéndose una disminución de su concentración sérica total, manteniéndose la concentración libre.
Intervalo de administración y momento de obtención de la muestra
Si el intervalo de administración de un antiepiléptico de eliminación rápida (valproico, primidona, carbamacepina) es mayor, las fluctuaciones demasiado amplias de su
concentración sérica pueden causar un "pico" demasiado alto, con fenómenos tóxicos transitorios o un "valle" menor, con disminución de la eficacia terapéutica.
Cumplimiento de la prescripción
El tratamiento antiepiléptico es crónico y con gran frecuencia no se toma correctamente la medicación, lo que impide que se alcancen los niveles séricos esperados para una determinada dosis16.
Índice nivel/dosis
Es un método sencillo para valorar cuantitativamente la relación dosis-nivel sérico alcanzado. Se calcula dividiendo el nivel estable (en mg/l) alcanzado en la fase de equilibrio estacionario entre la dosis (en mg/kg). Este índice nos da el nivel sérico hipotético alcanzado con una dosis teórica de 1 mg/kg y se utiliza para el ajuste de dosis y detección del incumplimiento en los fármacos con cinética lineal.
El índice nivel/dosis (N/D) varía entre distintos individuos y poblaciones, en función de distintos factores (edad, politerapia e incorrecto cumplimiento de la prescripción). En los niños aumenta con la edad hasta llegar a la adolescencia, donde alcanza los valores del adulto. En monoterapia es mayor que en politerapia, por la interacción con otros antiepilépticos inductores enzimáticos (como fenobarbital, fenitoína, carbamacepina, primidona) que aumentan la eliminación del antiepiléptico.
Aunque se han publicado diversos estudios sobre los valores del N/D de los antiepilépticos que difieren entre sí, en la tabla 3 se muestra una propuesta pragmática empírica de estos valores en monoterapia, según la edad de los pacientes17-20.

DIRECTRICES DE REFERENCIA
La utilización correcta de la TDM supone el cumplimiento de unos requisitos: idealmente, el motivo de la petición debe estar justificado, la extracción debe realizarse en el momento correcto, el resultado analítico debe ser preciso y exacto, interpretado correctamente y repercutir en la toma de decisiones terapéuticas adecuadas21.
Una limitación importante en la efectividad de la TDM es el alto porcentaje de peticiones sin motivo justificado y extracciones no realizadas en el momento correcto. Este último parece ser el motivo más frecuente de la incorrecta utilización de la TDM22,23.
Para facilitar su utilización correcta se han elaborado varias directrices (guidelines) sobre varios aspectos de la TDM de antiepilépticos21,24,25. Los métodos utilizados (cómo debe hacerse), los motivos de petición justificados (por qué debe hacerse) y el momento correcto de la extracción (cuándo debe hacerse).
Indicaciones de la monitorización
Motivos de petición justificados
Las principales indicaciones de la TDM de antiepilépticos reconocidas actualmente son:
Control del cumplimiento de la prescripción. Suele aceptarse que un 30% al 50% de los pacientes epilépticos no toman correctamente la medicación. El hallazgo de una concentración baja del fármaco, imprevisible para la dosis prescrita, o una ausencia del mismo, permite diagnosticar el mal cumplimiento o el incumplimiento de la prescripción16.
El N/D del fármaco orienta sobre el nivel previsible para la dosis prescrita y características del paciente (edad, monoterapia o tipo de tratamiento asociado). Cuando se han realizado varios controles al paciente podemos estimar el valor del N/D individual. La disminución clara del N/D de un paciente (> 20% en un control analítico) sugiere un cumplimiento incorrecto de la prescripción.
Individualización de la dosis. Debe prescribirse a cada individuo la pauta posológica que precisa según sus características (genéticas, fisiológicas, patológicas y farmacológicas) y que resulta más eficaz y segura. El objetivo sería conocer el nivel diana para cada paciente, con el que se controlan las crisis sin aparecer reacciones adversas. Este nivel es útil para controlar el cumplimiento y ajustar la dosis y puede variar según la situación clínica, por ejemplo, al agravarse la enfermedad epiléptica.
Para ajustar la dosis a dichas características pueden utilizarse tres métodos: prueba de ensayo y error, iniciando el tratamiento a dosis bajas, que se aumentan progresivamente hasta el control de las crisis o la aparición de efectos indeseables; utilización de nomogramas basados en parámetros farmacocinéticos poblacionales y el control de los niveles séricos de los fármacos una vez conseguida la fase de equilibrio estacionario.
La dosis inicial del antiepiléptico se elige en base a datos poblacionales del N/D. El objetivo inicial es conseguir un nivel estable próximo al límite inferior del rango terapéutico o intervalo óptimo. Posteriormente, si se consigue un buen control clínico de las crisis, se deben realizar controles periódicos (semestrales) para valorar el grado de cumplimiento y ajustar la dosis si fuese necesario (por aumento de peso en niños, enfermedad intercurrente o adición de un fármaco con interacción previsible). Los ajustes de dosis de fenitoína, por su cinética no lineal, pueden realizarse con ayuda de nomogramas26.
Si no se consiguiese inicialmente controlar las crisis, puede aumentarse la dosis gradualmente hasta alcanzar el límite terapéutico superior o la aparición de toxicidad, cambiar de antiepiléptico o añadir un segundo antiepiléptico.
Ineficacia del tratamiento. Puede producirse desde el comienzo del tratamiento o tras un período de control de las crisis. Una de sus causas es que no se hayan alcanzado concentraciones séricas eficaces del fármaco, que puede subsanarse mediante el ajuste de las dosis. Conviene recordar que tanto las concentraciones bajas como las altas pueden asociarse con la aparición o aumento de las crisis5.
Dicha ineficacia puede deberse a: la propia enfermedad (tiempo transcurrido sin tratamiento, aparición de las crisis a una edad temprana, gravedad de la epilepsia, asociación a trastornos psiquiátricos o neurológicos graves)27; al paciente (cumplimiento incorrecto o incumplimiento de la prescripción); al médico (diagnóstico incorrecto, elección incorrecta del antiepiléptico, dosis inadecuada del mismo, cambios frecuentes de la medicación).
Aparición de reacciones adversas o signos de toxicidad28,29. Es más frecuente con los antiepilépticos de bajo índice terapéutico (fenitoína, carbamacepina) y en situaciones de valoración clínica difícil (crisis infrecuentes, uso profiláctico de anticonvulsivantes). En la aparición de signos tóxicos en régimen de politerapia influye la sinergia aditiva entre los efectos indeseables de dos antiepilépticos (por ejemplo, entre carbamacepina y fenitoína o fenobarbital)30 y la posible sobredosificación del fármaco (absoluta o relativa). En la sobredosificación relativa pueden
influir las interacciones con otros fármacos o las características individuales, por ejemplo, menor eliminación.
Si el efecto indeseable o los signos de toxicidad son graves hay que dar preferencia a su control antes que al de las crisis.
La TDM también facilita el diagnóstico y prevención de efectos indeseables en niños pequeños o deficientes mentales, siempre difícil.
Entre las reacciones adversas al tratamiento antiepiléptico no relacionadas con la dosis o la concentración sérica se encuentran:
Reacciones inmunológicas: fiebre, erupciones cutáneas (exantema, síndrome de Stevens-Johnson, dermatitis exfoliativa, síndrome de Lyell), alteraciones hematológicas (trombocitopenia, neutropenia o agranulocitosis, eosinofilia, anemia aplásica), hepatitis y lupus eritematoso sistémico.
Efectos adversos diferidos: diversas malformaciones congénitas (craneofaciales, digitales, labio leporino y fisura palatina, cardiopatía congénita, microcefalia, defectos del tubo neural) se han asociado al tratamiento con varios antiepilépticos. El riesgo de teratogénesis es mayor en politerapia y deben valorarse varias opciones terapéuticas en la mujer epiléptica embarazada: valorar la disminución gradual de dosis para intentar suspender temporalmente el antiepiléptico durante el primer trimestre de gestación si la mujer no hubiese tenido crisis en los 2 años previos al embarazo; intentar la monoterapia a las menores dosis eficaces; evitar los cambios de tratamiento; administrar suplementos de ácido fólico durante el primer trimestre, para prevenir el riesgo de defectos del tubo neural.
La utilidad de la TDM para detectar o prevenir reacciones adversas está limitada exclusivamente a 3 casos: intoxicaciones agudas por sobredosificación (tabla 4); efectos tóxicos sobre el sistema nervioso central (tabla 5) y reacciones predecibles, relacionadas con la dosis o el nivel sérico, en el tratamiento crónico. En estos casos, la disminución de la dosis y/o la suspensión temporal del tratamiento suelen resolver la situación. En ocasiones puede ser beneficioso aumentar el número de tomas diarias, para evitar "picos" altos de concentraciones séricas.


Los efectos tóxicos de los antiepilépticos sobre el sistema nervioso central son frecuentes, predecibles, relacionados con la dosis y controlables con individualización del tratamiento. La mayoría de antiepilépticos producen estos efectos. Hay una mayor especificidad por algunos de estos efectos adversos (por ejemplo, la fenitoína por alteraciones cerebelo-vestibulares; el fenobarbital por las alteraciones del sistema reticular ascendente). Pero en presencia de una intoxicación es difícil atribuir a uno de ellos la intoxicación.
Hay un tipo de toxicidad crónica que interfiere con el rendimiento psicomotor (por ejemplo, las alteraciones por fenobarbital que reducen la integración social del paciente). Puede relacionarse con la dosis y beneficiarse de una disminución de la misma.
Los niveles asociados con toxicidad son variables y no existe una concentración mínima tóxica aplicable a todos los individuos. Sin embargo, la asociación de niveles superiores al intervalo óptimo de concentraciones con síntomas o signos sospechosos de toxicidad permite atribuir ésta al antiepiléptico. En estos casos se aconseja disminuir la dosis tras omitir una o dos tomas; la fenitoína puede requerir suspender el tratamiento durante 1-3 días, comprobando la disminución de la concentración hasta niveles óptimos antes de reiniciar la administración.
Para la realización de ensayos clínicos es conveniente la determinación cuantitativa del compuesto estudiado, para conocer su farmacocinética y la relación entre sus concentraciones séricas y sus efectos terapéuticos y tóxicos.
Momentos de la monitorización
Son los siguientes: tras iniciar el tratamiento antiepiléptico; tras cambio de la dosis o pauta de administración; tras añadir un segundo antiepiléptico, con interacción previsible; tras un cambio en la función hepática, cardíaca o gastrointestinal; y en la supervisión periódica (semestral) del cumplimiento de la prescripción.
Indicaciones para determinar concentraciones libres
Esta determinación es potencialmente útil para aquellos antiepilépticos con alto grado de unión a las proteínas séricas, en los que un cambio en la fracción libre (concentración sérica libre/concentración sérica total) produciría una variación proporcionalmente mayor en la concentración sérica libre, que podría ocasionar cambios en la eficacia del fármaco31.
Actualmente, se acepta determinar la concentración libre de fenitoína o valproico en los pacientes y situaciones siguientes: concentración terapéutica del fármaco y toxicidad inesperada; paciente anciano, embarazada o neonato; paciente urémico; concentración de albúmina < 377 µmol/l (< 2.5 g/dl); y tratamiento asociado con fármacos que compiten por sitios de unión a proteínas (por ejemplo, asociaciones de ácido valproico, salicilato, fenitoína).
La determinación de rutina de la concentración libre del resto de antiepilépticos no es necesaria, porque las concentraciones total y libre se correlacionan bien y esta última es más costosa y complicada técnicamente.
Indicaciones para determinar metabolitos activos
La concentración alcanzada por los metabolitos activos de los antiepilépticos puede influir en su eficacia clínica. Concretamente, en el caso de la primidona es imprescindible determinar la concentración de fenobarbital, metabolito activo de aquélla; su determinación proporciona un mejor índice de eficacia o seguridad que la propia primidona, por tener menos exigencias que el fármaco original respecto al momento de la extracción (la fluctuación de las concentraciones de fenobarbital en el intervalo de administración es escasa, por su eliminación lenta).
Además se aconseja determinar el epóxido de carbamacepina en los casos en que puede aumentar su concentración sérica, como en los tratamientos asociados convalproico, fenitoína, fenobarbital y algunos nuevos antiepilépticos (lamotrigina y felbamato).
Momento de la extracción
Los momentos idóneos son:
En el "valle" (antes de la toma de la medicación), lo que es más importante para fármacos de eliminación rápida y vida media corta (carbamacepina, valproico, primidona) porque su concentración fluctúa ampliamente durante el intervalo de administración; o 1-4 horas tras la administración de una dosis de carga de fenitoína intravenosa.
Una vez conseguido el nivel estable (equilibrio estacionario), lo que sucede tras 4-5 vidas medias siguiendo una misma pauta de tratamiento; este tiempo es de 30 días (fenobarbital, carbamacepina), 21 días (fenitoína), 4 días (valproico), 2 días (primidona). En todo caso, es aconsejable esperar que pase un mes con la misma pauta de tratamiento, para que se alcancen el nivel estable de fenobarbital (metabolito de primidona), el grado máximo de autoinducción del metabolismo de la carbamacepina y la eficacia derivada de los metabolitos de valproico.
En una hora del día similar para el mismo paciente (las concentraciones séricas de carbamacepina y valproico son influidas por un ritmo circadiano).
También puede hacerse la extracción, aunque no se hayan extraído las muestras en la fase de equilibrio estacionario y el nivel "valle", en los siguientes casos: a) en las 6 horas siguientes a una crisis en un paciente previamente controlado; b) tras un empeoramiento inexplicado en el control clínico de las crisis, que podría asociarse con niveles altos o bajos y se necesita conocerlos para ajustar la dosis; c) en sospecha de toxicidad relacionada con la dosis, para evitar una nueva administración del antiepiléptico en caso de que hubiese niveles altos que permitiesen atribuir la toxicidad al fármaco, y d) en sospecha de mal cumplimiento de la prescripción, que se diagnosticaría con una concentración casi nula o desproporcionadamente baja sin relación con la dosis prescrita.
Es importante anotar en la hoja de petición los momentos de la última toma del fármaco y la extracción, así como el tiempo de tratamiento con el fármaco a la misma pauta.
Tipo de muestra
Matriz
En general se emplea el suero, que es imprescindible si va a medirse la concentración sérica libre de un antiepiléptico, ya que algunos anticoagulantes interfieren con la unión a proteínas. Otros líquidos biológicos que han sido probados son la saliva y las lágrimas, porque su pobreza en proteínas podría hacerlos reflejar la concentración libre de algunos antiepilépticos.
Volumen de muestra
De 2 a 5 ml de sangre sin ningún anticoagulante, extraída por punción de una vena sin vía o proximal a la vía, para evitar resultados erróneos por contaminación o dilución. En neonatos son suficientes muestras de capilares sin anticoagulantes, extraídas por punción de un dedo o del talón.
Tubos de extracción
Generalmente se recomienda evitar el uso de: anticoagulantes con citrato y oxalato (disminuyen la concentración sérica total de fenitoína y valproico); heparina, porque aumenta la fracción libre (activa las lipoproteinlipasas, lo que produce un aumento de ácidos grasos libres, que desplazan al antiepiléptico de su unión a la albúmina); tubos con separador de suero (la absorbancia del fármaco in vitro puede disminuir las concentraciones séricas de fenitoína, siendo conveniente rellenar los tubos completamente y procesar y extraer el suero rápidamente); EDTA, porque su efecto sobre las concentraciones de antiepilépticos no está bien establecido.
Precisión analítica
Las necesidades de precisión analítica dependen de:
Diferencia o margen de seguridad entre las concentraciones séricas del límite terapéutico superior y las mínimas asociadas con toxicidad. Este margen es variable entre los antiepilépticos, siendo mayor para valproico, pero a veces es inexistente, porque en algunos pacientes puede aparecer la toxicidad con concentraciones situadas dentro del rango terapéutico.
Proporción existente entre los cambios mínimos de dosificación que pueden realizarse y la dosis prescrita. Estos cambios dependen de las presentaciones de menor dosis que existan en el mercado farmacéutico. En los antiepilépticos, sólo pueden hacerse ajustes finos de la dosis con valproico y fenobarbital, lo que limita la exigencia de precisión analítica marcada por el escaso margen de seguridad de estos fármacos.
En las determinaciones de la concentración sérica total de antiepilépticos, unos resultados de ± 10% del resultado actual son aceptables, probablemente, para todos los antiepilépticos. Esto significa que se requiere una precisión interanálisis con un coeficiente de variación del 5%-10%, o menor.
Puede haber una mayor variabilidad en los resultados de la concentración sérica libre de fenitoína para una muestra determinada, debido a un mal control de la temperatura durante la fase de ultrafiltración.
VENTAJAS Y UTILIDAD
La TDM puede aumentar la efectividad del tratamiento antiepiléptico, si contribuye a lograr un mejor control de las crisis o la detección de síntomas o signos tóxicos. Así, puede detectar concentraciones altas o bajas, un cumplimiento incorrecto de la prescripción o interacciones farmacológicas. En caso de que se hayan logrado concentraciones óptimas y persistan las crisis, la TDM indica si no es oportuno seguir el tratamiento con ese mismo fármaco.
También puede identificar pacientes o subgrupos de población de características clínicas y farmacocinéticas particulares con trascendencia terapéutica32. Finalmente, permite elaborar bases de datos con las características clínicas, cinéticas y terapéuticas de los pacientes, de interés epidemiológico33.
MONITORIZACION TERAPÉUTICA DE LOS NUEVOS ANTIEPILÉPTICOS
Dicha monitorización no es aplicable a todos los pacientes ni tiene unas directrices tan claras como ocurre con los antiepilépticos clásicos, porque los nuevos antiepilépticos son mejor tolerados y presentan menos interacciones que aquéllos.
Los intervalos óptimos de niveles de los nuevos antiepilépticos están escasamente definidos, por varios motivos: ausencia de técnicas analíticas sencillas disponibles; escasez de estudios; suelen utilizarse como tratamiento añadido a pacientes epilépticos resistentes.
La monitorización de niveles séricos puede tener interés para lamotrigina, topiramato, oxcarbacepina, tiagabina, zonisamida y felbamato34. Sin embargo, esta monitorización está en fase de desarrollo y tiene utilidad en unidades especializadas, tanto para individualizar la dosis, como para conocer mejor las relaciones dosis-nivel y nivel-efecto. Es previsible que el mejor conocimiento de la farmacocinética clínica y la monitorización de niveles de los nuevos antiepilépticos mejore su utilización terapéutica8.
